Home > D. General pathology > Blood and immunity > Dysimmune diseases > chronic granulomatous disease
chronic granulomatous disease
MIM.306400
Friday 3 October 2003
Digital cases
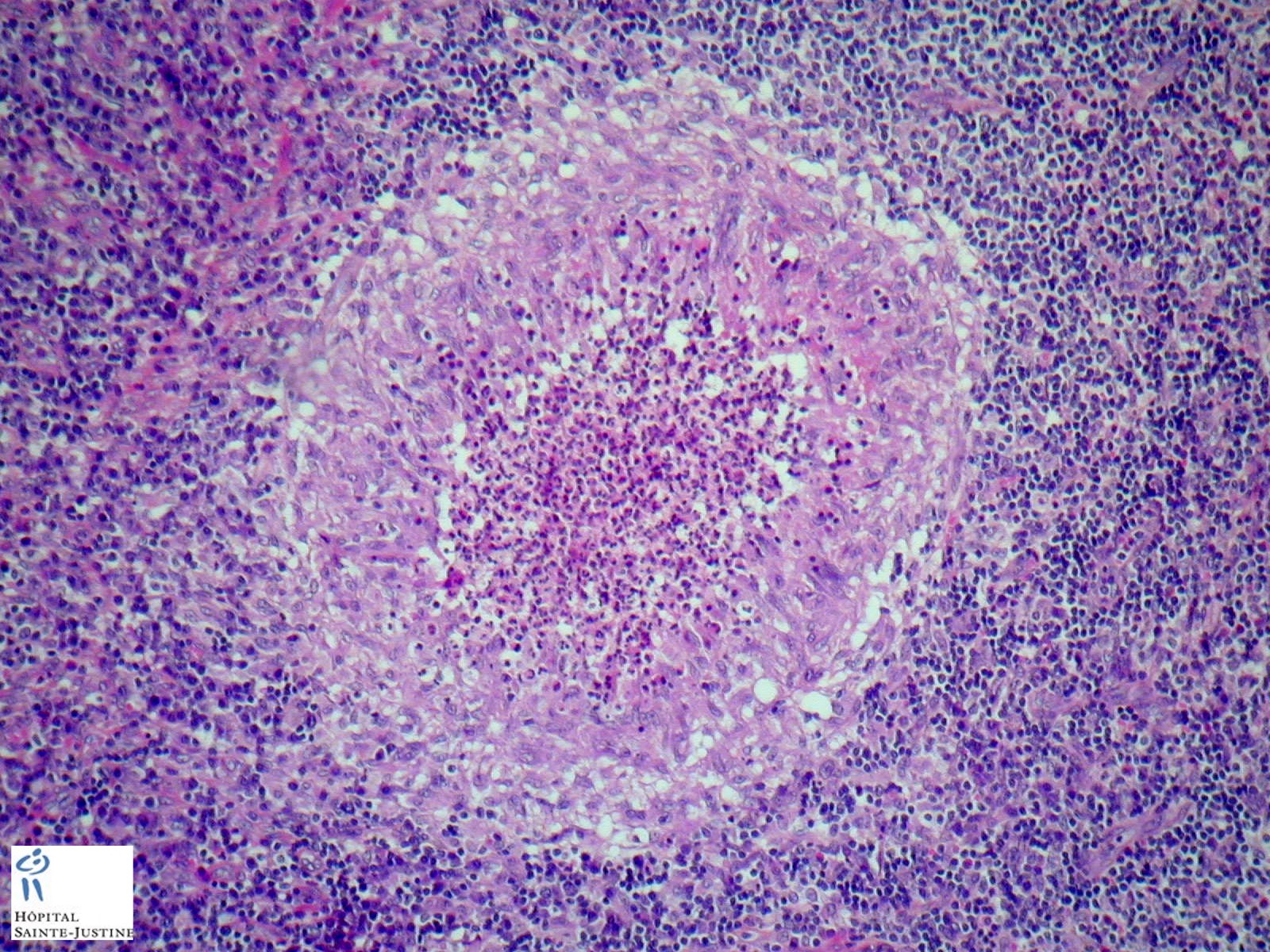
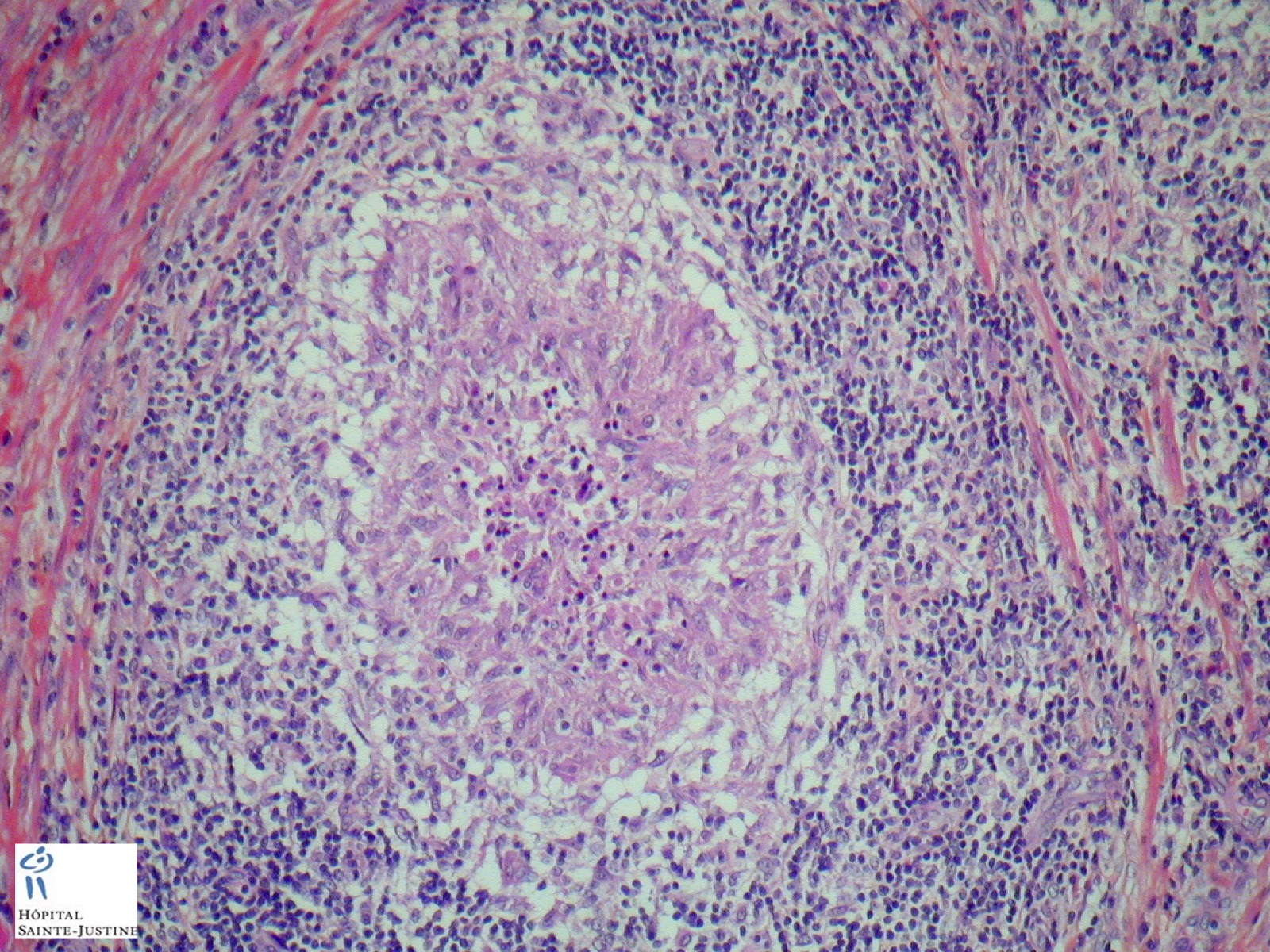
Case 20 : Foamy macrophages in portal spaces - Chronic granulomatous disease
Case 252 : Cerebral granulomatosis in chronic granulomatous disease (CGD)
Definition: chronic granulomatous disease (CGD) is a hereditary disease where neutrophil granulocytes are unable to destroy ingested pathogens. It leads to the formation of granulomata in many organs (granulomatosis). This rare disease occurs in about 1 on 200,000 - 250,000 live births.
The continuous development of repetitive infections ranging from pneumonia, to meningitis, to multisystemic aspergillosis, associated with failure to thrive and characteristic granulomatous lesions is the classic clinical presentation of CGD.
Its estimated incidence is between 1 in 200 000 in the US and 1 in 450 000 in Sweden.
Chronic Granulomatous Disease (CGD) is a genetic disorder causing dysfunction in the NADPH oxidase complex of phagocytic cells, which decreases greatly their bactericidal and fungicidal properties.
Chronic Granulomatous Disease (CGD) can be associated with frequent and severe infections, granulomas and inflammation in many organs, causing lesional syndromes as colitis or pneumonitis.
Synopsis
frequent and severe infections
granulomas and inflammation in many organs
colitis
- “Crohn-like” ulcerated granulomatous ileocolitis
nodal epitheloid granulomas with giant cells and caseous necrosis
hepatic non-epitheloid granulomas
recurring perianal abcesses and fistulae
granulomatous pneumonitis
granulomatous bronchitis
granulomatous adenitis
non caseating non-specific inflammatory granulomas
Staphylococcus aureus hepatic abscess
severe recurrent bacterial infections
- Serratia marcescens
infectious lymphadenitis (infectious adenitis)
- bacteria lymphadenitis adenitis
- S. aureus
- mycobacteria
pulmonary infection due to Aspergillus nidulans
fungal osteomyelitis
fungal lymphadenitis
- Candida sp. (Candida albicans, Candida glabrata)
- Candida glabrata granulomatous lymphadenitis with caseous necrosis
chronic disseminated candidiasis (CDC or hepatosplenic candidiasis)
nodal infections (infectious adenitis)
- bacterial adenitis
- Staphylococcus aureus
- Enterobacteriacea
- gram-positive bacilli: Nocardia sp.
- Granulibacter besthedensis (necrotizing adenitis)
- mycobacteria lymphadenitis
- Mycobacterium bovis (BCG)
Aspergillus sp. is the most frequently encountered fungus in chronic granulomatous disease (CGD), but Candida sp. is sometimes associated with infections in CGD (7.5% of all fungal infections).
Candida sp. infections can manifest as subcutaneous abscesses, candidemia, pneumonia, meningitis or occasionally suppurative adenitis.
CDC is associated with non caseating granulomas involving the liver, spleen and occasionally kidneys or lungs.
Physiopathology
CGD represents the archetype of inflammatory cellular phagocytic defects involving intracellular microbicidal mechanisms.
The specific impairment lies on an enzymatic complex responsible for producing hydrogen peroxide: the NADPH oxidase. This system is responsible for the generation of superoxide, the precursor of several oxidant molecules necessary for adequate intracellular processing of phagocytized microbes.
The enzymatic complex is most abundant in neutrophils, eosinophils and monocyte/macrophage cells, and it is composed of a membrane-bound flavocytochrome ß558 and four cytosolic proteins p47phox, p67phox, p40phox , and p21rac. Seventy percent of patients with CGD inherit the disorder on an X-linked pattern with a mutated gp91phox, the gene responsible for encoding the ßsubunit of the flavocytochrome.
The second most common mutation involves p47phox, inherited in an autosomal recessive pattern.
Prenatal diagnosis may be established by analysis of neutrophils obtained from umbilical vein samples through fetoscopy; DNA from amniocytes and chorionic villous sampling may also be used.
Patients with CGD are usually infected by catalase-positive micro-organisms, most frequently Aspergillus sp., S. aureus, Serratia sp., Nocardia sp. and B. cepacia.
Catalase degrades the small amounts of H2O2 produced by the infecting microbes, allowing the them to survive within the phagosome.
Catalase-negative microorganisms apparently generate and maintain sufficient amounts of H2O2 from which other highly reactive compounds are produced within the phagocytic vacuole of the CGD neutrophils and macrophages, leading to effective microbial killing.
Hence, these microorganisms are usually not seen in CGD patients.Death is most commonly associated with Aspergillus infection.
CGD patients may have muffled symptoms even when undergoing severe infections. Fever, leukocytosis and other indications of their septic state may be absent or subtle, which warrants a careful monitoring.
Additional complications of CGD patients are related to exuberant and persistent granulomatous inflammation, with extensive skin ulceration, systemic lupus erythematosus-like disease, pneumonitis, Crohn’s-like disease, gastrointestinal and genitourinary tract obstruction.
Long-term antibiotic therapy, steroids, interferon-_, granulocyte transfusions, bone marrow transplantation and gene therapy, all have important roles in the adequate treatment of these patients.
Synopsis
systemic granulomatosis
- tuberculoid granuloma with central suppuration
infections
- catalase + bacterias
- mycobactetioses (17544093)
- BCG-osis
- BCG-itis
- tuberculosis (Mycobacterium tuberculosis)
inflammatory pseudotumor
- bladder (veiscal inflammatory pseudotumor) (11956865)
Localization
cutaneous CGD
digestive CGD
- colonic CGD
hepatic CGD (CGD in the liver)
pulmonary CGD
Etiology
Four genes have been implicated in CGD:
CYBB, coding the gp91-phox subunit (X-linked, accounts for 2/3 of the cases);
CYBA, coding p22-phox;
NCF1, coding p47-phox;
NCF2, coding p67-phox
cytochrome b-negative CGD
- X-linked cytochrome b-negative CGD
- CYBB (Xp21.1): X-linked CGD (MIM.306400) is caused by mutation in the gene encoding p91-phox (CYBB) (MIM.300481) (Xp21.1)
- autosomal recessive cytochrome b-negative CGD
- CYBA: mutations of CYBA in autosomal recessive cytochrome b-negative CGD (MIM.233690)
cytochrome b-positive CGD
-* NCF1 (7q11.23): autosomal recessive cytochrome b-positive CGD type 1 (MIM.233700) is caused by mutation in the NCF1 gene (MIM.608512), which encodes the p47-phox (phagocyte oxidase) protein.
-
- Neutrophil cytosolic factor-1 (NCF1) is a component of the NADPH oxidase complex.
NCF2 (1q25): autosomal cytochrome b-positive CGD type 2 (MIM.608515) is caused by mutation in the NCF2 gene (MIM.233710), which encodes the p67-phox protein (1q25).
- Neutrophil cytosolic factor-2 (NCF2) is a component of the NADPH oxidase complex.
A fifth gene, coding for p40-phox, has not been implicated
A low level of NADPH, the cofactor required for superoxide synthesis, can lead to CGD. This has been reported in women who are homozygous for the genetic defect causing glucose-6-phosphate dehydrogenase deficiency (G6PD), which is characterised by reduced NADPH levels.
Prognosis
Survival of CGD patients has improved from only 21% of patients living beyond 5 years in 1967, to 92% reaching 8 years of age.
In a report of patients born over a 32-year period, 50% survived through the third decade of live.
The mean age of survivors in 1998 was 16 years.
Difficulties with compliance of therapy in adolescents are thought to be responsible for the failure to improve the overall mortality rate.
See also
granulomatoses
genetic immune deficiencies
leukocyte oxidant metabolism
References
Zambrano E, Esper F, Rosenberg R, Kim J, Reyes-Mugica M. Chronic granulomatous disease. Pediatr Dev Pathol. 2003 Nov-Dec;6(6):577-81. PMID: 15018460
Heyworth PG, Cross AR, Curnutte JT. Chronic granulomatous disease. Curr Opin Immunol 2003;15:578-584.