Home > E. Pathology by systems > Urinary system > Kidneys > clear cell sarcoma of the kidney
clear cell sarcoma of the kidney
Wednesday 14 July 2004, by
CCSK, renal clear cell sarcoma; bone-metastasizing renal tumour of childhood
| AGCOH | PathConsult | eMedicine |
Digital slides
Case 66 (HPC:66) : Clear cell sarcoma of the kidney (CCSK)
Definition: Clear cell sarcoma of the kidney (CCSK) is a malignant renal tumor of childhood with a propensity to metastasize to bone and other organs. CCSK is linked to internal tandem duplications (ITDs) of the BCOR gene.
This tumor may also recur many years after its initial diagnosis. The average age at diagnosis is 2-4 years.
CCSK is unrelated to the clear cell sarcoma of the soft tissue, also known as malignant melanoma of soft parts. Extrarenal tumors histologically identical to CCSK have been reported in rare instances.
This tumor may be confused with other pediatric renal tumors including blastema-predominant Wilms’ tumor, malignant rhabdoid tumor, and cellular mesoblastic nephroma.
Epidemiology
CCSK comprises 5 percent of primary pediatric renal tumors with the peak incidence in the second year of life; however, patients’ ages have ranged from 2 months to 54 years. Adult cases are extraordinarily rare.
CCSK does not appear to be associated with genetic syndromes like Wilms tumor (i.e. WAGR syndrome, Beckwith-Wiedemann syndrome, and Denys-Drash syndromes).
Males appear to be more commonly affected than females.
The National Wilms’ Tumor Study (NWTS) has classified CCSK as one of the most common of the prognostically unfavorable histology tumors (others being anaplastic Wilms’ tumor and malignant rhabdoid tumor of the kidney).
The age at presentation for NWTS cases ranged from 2 months to 14 years, with a mean age of 36 months.
Among 351 total patients in the NWTS study, the highest incidence of CCSK was in years 2 and 3 of life in which 50% of cases were diagnosed. A sharp decline occurred thereafter.
A male predominance was noted: there were 231 males and 113 females (ratio: 2.04:1) in the study.
Only one CCSK among the 351 reviewed was associated with perilobar nephrogenic rests. No case was associated with intralobar nephrogenic rests.
Only a single CCSK was associated with renal dysplasia.
No familial or syndrome-associated CCSKs have been identified. However, the brother of one patient with CCSK in NWTS study subsequently developed a nephrogenic (metanephric) adenofibroma, another rare renal neoplasm with a stromal component.
Clinics
The usual presentation of CCSK is a child with a flank mass with or without hematuria much like the typical signs and symptoms associated with Wilms’ tumor. Abdominal pain and fever may also occur. In some instances, patients present with pathologic fractures due to metastatic tumor.
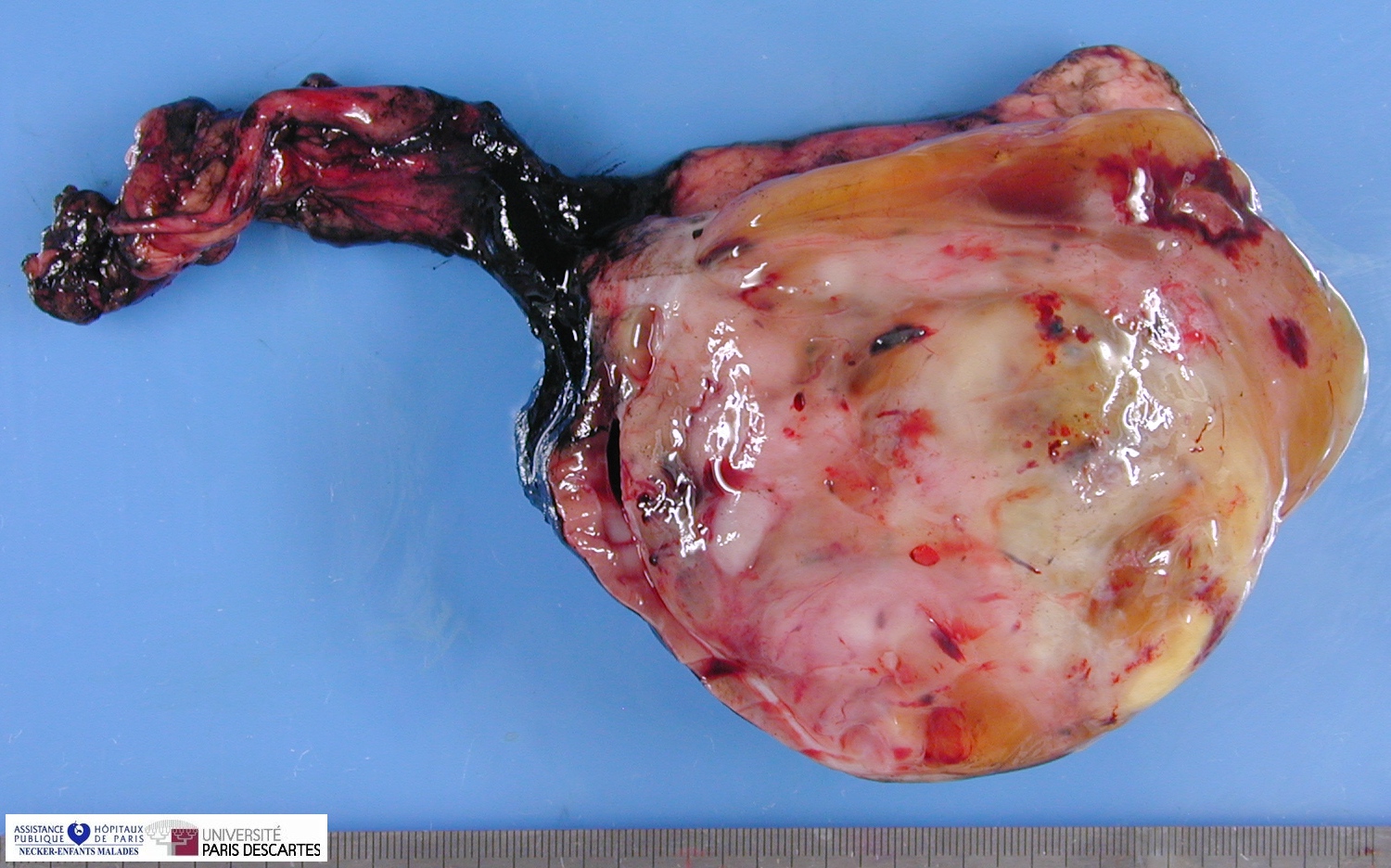
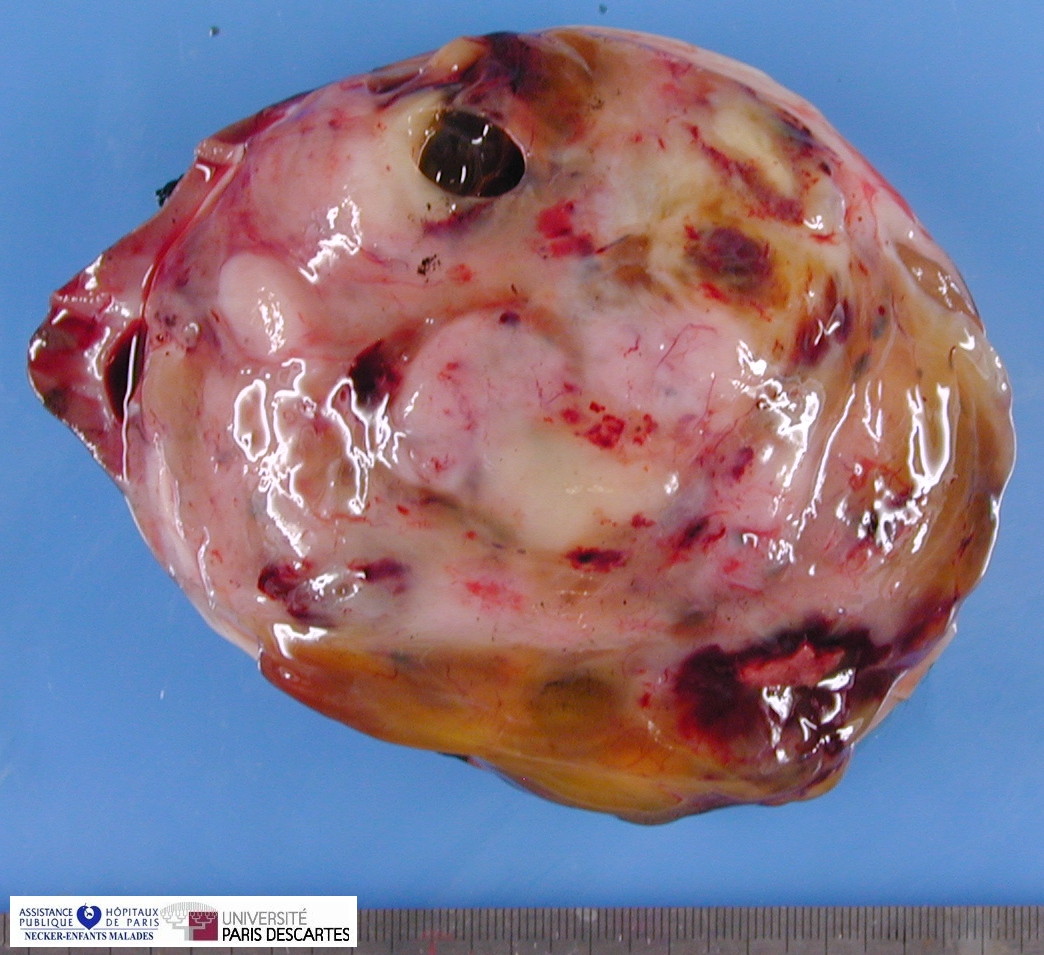
Macroscopy
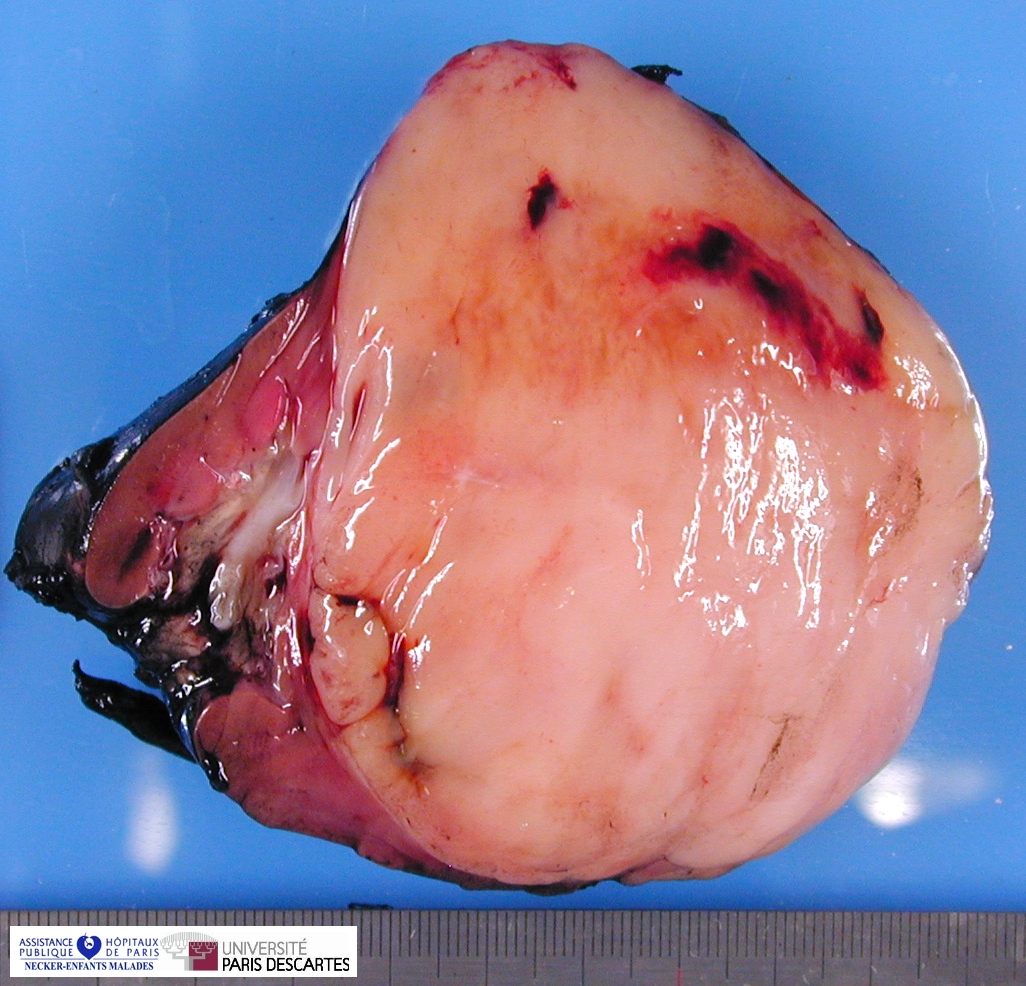
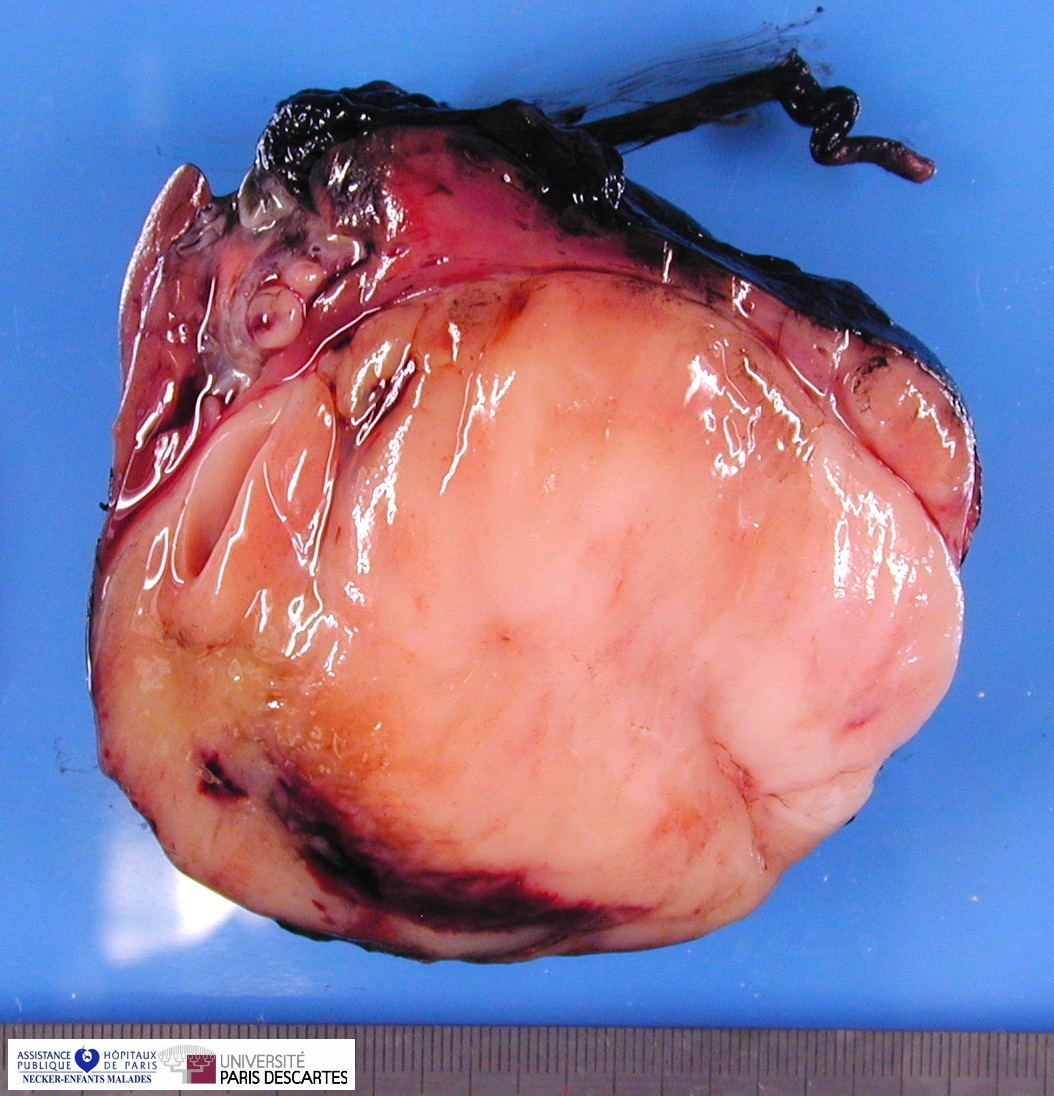
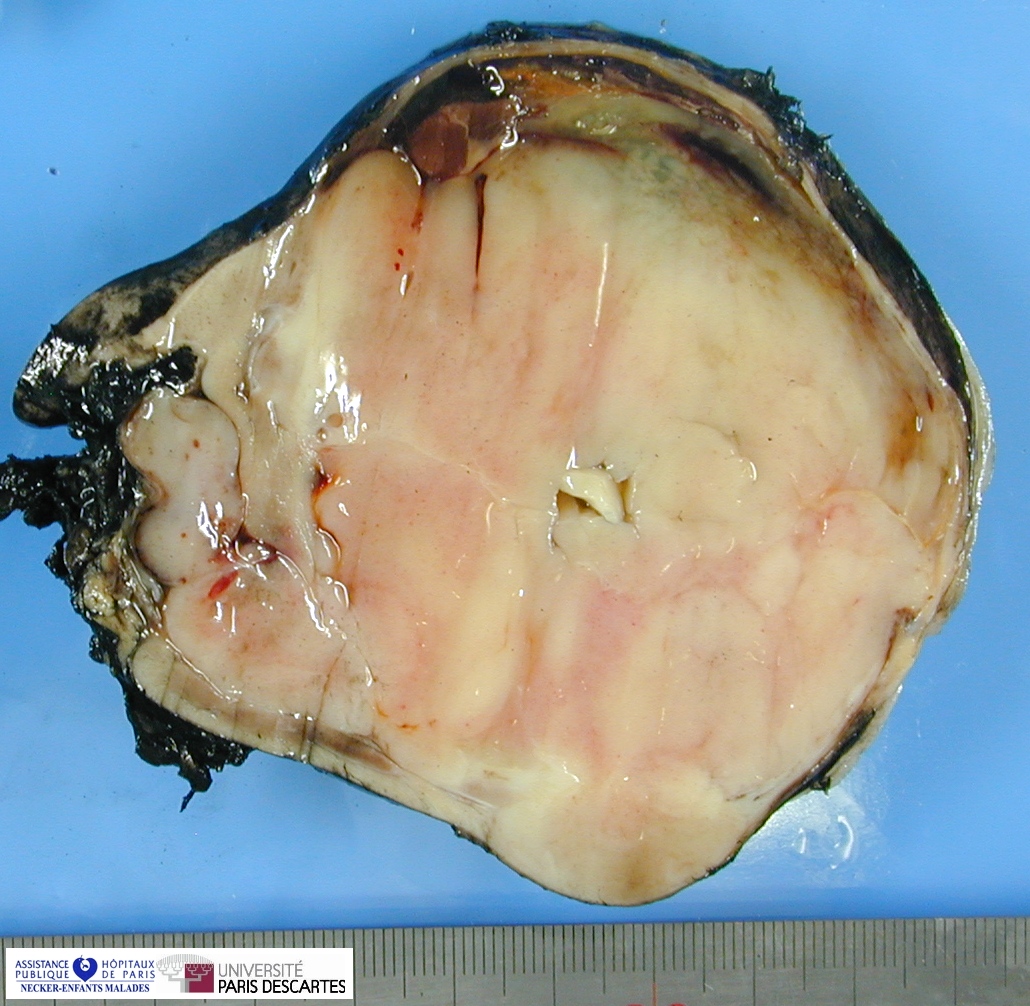
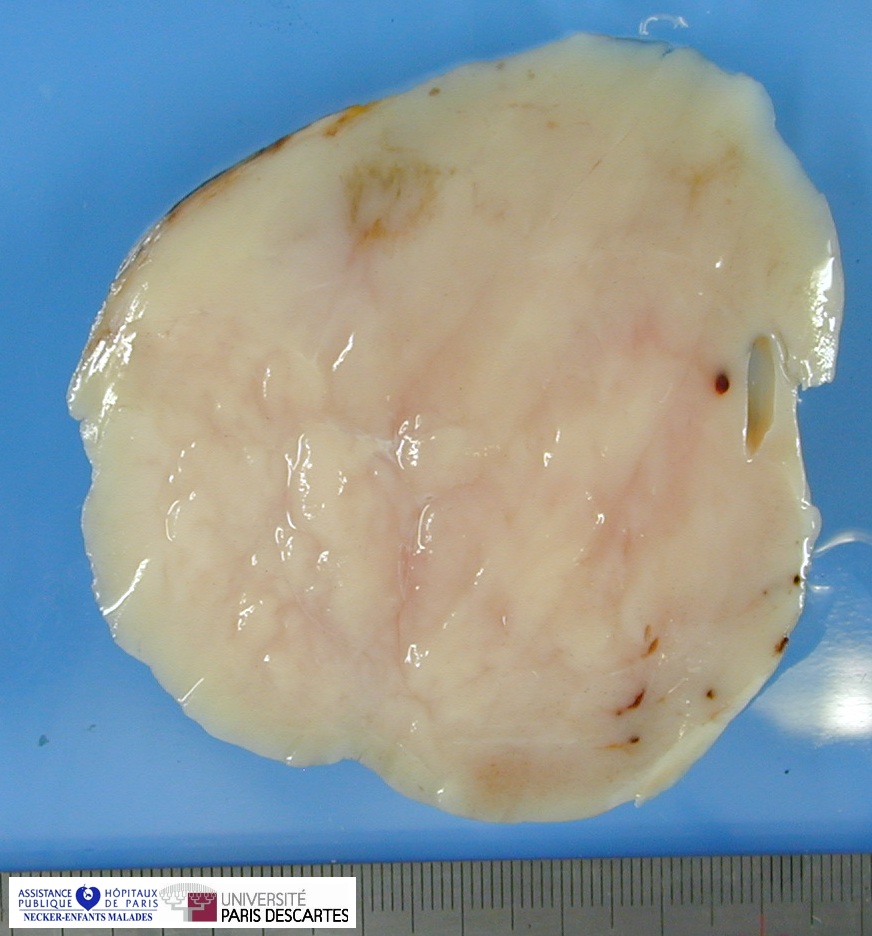
Grossly, the tumor arises within the renal medulla has a mass of up to 3,000 grams.
On cut section, the tumor is usually white-tan to gray and has a firm texture and is sharply defined from the surrounding renal parenchyma.
CCSK usually presented as a large, unicentric mass markedly distorting or nearly completely replacing the kidney; the mean diameter of measured tumors was 11.3 cm (range, 2.3–24 cm).
The mean of the more consistently measured combined kidney–tumor specimen weight was 661 g (range, 43.5–1950 g). When an epicenter could be determined, the renal medulla was the most common location. No case of multicentric origin was identified.
On cut section, tumors were most commonly described as tan–grey, soft, and mucoid. Cystic foci were nearly universal and occasionally represented the dominant feature such that a radiologic and gross pathologic diagnosis of multilocular renal cyst (cystic nephroma) was made. The microscopic appearance of such lesions also mimicked cystic nephroma.
Within these typically large and relatively homogeneous tumors, discrete foci of necrosis (73% of cases) and hemorrhage were common. A subset of tumors had a more firm, whorled appearance.
Tumors usually appeared grossly well-circumscribed with a sharp kidney–tumor border. Gross extension into the renal vein (tumor thrombus) was evident in 5% of cases.
Microscopy
Histologically, the classical CCSK (features present at least focally in over 90% of tumors) is composed of nests and cords of cells with scant cytoplasm and high nuclear-cytoplasmic ratios.
The tumor has a prominent vascular network that may be highlighted with Ulex Europeous I lectin or monoclonal antibodies specific for factor VIII or CD31.
Adundant collagenous (sclerotic) extracellular matrix material is also a common finding in classical CCSK. The nuclei are characterized by a fine chromatin pattern and mitotic figures are generally rarely identified.
Isolated nephrons are entrapped by the tumor.
Classic pattern
Classical pattern demonstrates nests (or cords) of ovoid cells separated by fibrovascular septa
- cord cells may be ovoid, epithelioid or spindled with bland nuclei without prominent nucleoli
- septa demonstrate marked regular branching ‘chicken wire’ pattern of capillaries
- uniform appearance of apparently bland, plump, ovoid cells at low power.
The classic histologic pattern of clear cell sarcoma of the kidney is characterized by cord cells arranged in cords, nests, or groups surrounded by thin fibrovascular septa.
A moderate amount of clear intercellular matrix separates the cord cells, giving a clear appearance, hence the designation clear cell sarcoma of the kidney.
The term clear cells is doubly a misnomer because the clear cell appearance is caused by loose spacing of the round or oval cord cells with intervening intercellular clear mucoid matrix and because the clear appearance may be absent in many cases.
Characteristic infiltrative border on high power (although the lesion may appear well circumscribed on low power).
Like other renal tumors of childhood, CCSK is staged by the National Wilms’ Tumor Study (NWTS) staging scheme.
The classic pattern of CCSK was defined by nests or cords of cells separated by regularly spaced, arborizing fibrovascular septa.
The regularity of the septal spacing resulted in cord widths of between four and 10 cells. While usually plump and ovoid, the cord cells not uncommonly assumed a spindle shape.
The septa ranged from thin, regularly branching “chicken-wire” capillaries highly reminiscent of the architecture of myxoid liposarcoma to capillaries enmeshed by sheaths of fibroblast-like cells set in a collagenous matrix.
These “cellular septa” measured up to 50 μm in width and are a characteristic feature of CCSK. Rarely, dilation of the thin septal capillaries created a staghorn, hemangiopericytomatous appearance.
The cord cells featured nuclei that were overall uniform in shape, although fine irregularities in contour could be appreciated at high magnification. The chromatin was characteristically of a fine, dusty texture without prominent nucleoli or coarse condensations. Empty-appearing “Orphan Annie” eye nuclei were frequent. These characteristic nuclear features were usually not evident on either original frozen sections or permanent sections prepared from previously frozen tissue, and hence are fixation-dependent.
The cord cells are usually loosely spaced, separated from their neighbors by optically clear material that has proven to be extracellular mucopolysaccharide matrix, and gives rise to the clear cell appearance.
Nuclear overlap was less common than in Wilms tumor, PNET, or other more cellular pediatric renal neoplasms.
The cytoplasm was generally sparse with indistinct cell borders. However, well-demarcated pink cytoplasm was occasionally evident, particularly in areas of acinar-type epithelioid pattern or following fixation in mercuric fixatives such as B5.
The combination of focal cord cell crowding and acidophilic cytoplasm imparted a “dark cell” appearance. Rarely, rhabdoid cytoplasmic inclusions were identified, but these cells lacked the prominent nucleolus characteristic of true rhabdoid tumors.
While tumors appeared circumscribed both grossly and under low-power microscopic examination, on closer inspection they entrapped individual tubules of the medulla and occasionally cortical glomeruli.
Entrapped tubules often acquired a metaplastic “embryonal” appearance with occasional mitoses, simulating the neoplastic tubules of Wilms tumor.
Alternatively, the entrapped tubules dilated extensively, giving rise to grossly evident cysts with paucicellular septa. In rare cases, an exaggerated cystic appearance microscopically simulated cystic nephroma in the majority of the slides. Only focally did areas diagnostic of CCSK emerge.
Subtypes
Several histologic variants of CCSK are recognized.
While most tumors (91%) had the classic pattern as either a predominant or a secondary morphology, a majority also demonstrated one or more variant patterns. The variant patterns of CCSK can best be understood as alterations of either cord or septal cell morphology. This concept does not imply these cell types are biologically distinct.
These patterns usually blended smoothly with the classic pattern or another variant pattern, with two exceptions below. The variant patterns recognized and the percentage of cases in which they were seen are as follows:
1. Myxoid pattern (50%)
2. Sclerosing pattern (35%)
3. Cellular pattern (26%)
4. Epithelioid pattern (trabecular or acinar type) (13%)
5. Palisading (verocay-body) pattern (11%)
6. Spindle cell pattern (7%)
7. Storiform pattern (4%)
8. Anaplastic pattern (2.6%)
Myxoid pattern (50%)
- The most common variant is the myxoid CCSK.
- This histology features diffuse accumulation of mucopolysaccharide matrix material between tumor cells sometimes creating a cystic appearance.
- The myxoid pattern featured pools of amphophilic extracellular material separating the cord cells.
- This material ranged from the minute deposits to large grossly evident expanses of mucin creating large pseudocysts that were difficult to distinguish from the true cysts formed from entrapped tubules at the tumor periphery.
- The extracellular mucoid material possessed the histochemical staining characteristics of hyaluronic acid; whether between closely spaced cells or in large pools, it was light blue on alcian blue stain, and staining was sensitive to hyaluronidase treatment.
- The appearance of the cord cells at the periphery of these mucoid pools was variable. Some remained similar to their neighboring cells closer to the septa.
- Others acquired a more epithelioid appearance, simulating gland formation around the smaller pools. Others clung to the septa, creating an anastamosing channel pattern that simulated a vascular neoplasm.
Sclerosing pattern (35%)
- The sclerosing variant of CCSK is characterized by prominent collagen bundles that may isolate single or small groups of tumor cells in a dense matrix that may become hyalinized.
- The sclerosing pattern usually featured deposition of acellular, osteoid-like material between the septa and the cord cells. In this configuration, the cord cells became compressed on both sides by the hyaline material resulting in an Indian-file pattern.
- When the cord cell cytoplasm became attenuated and the nucleus was compressed against the extracellular hyaline material, the appearance mimicked the hyaline intracytoplasmic inclusions of RTK.
- Lesions acquired a paucicellular atrophic appearance when the sclerosis was less hyaline and more collagenous in nature.
- The sclerosing pattern appeared to arise from the myxoid pattern in some tumors in which the amphophilic extracellular material became progressively hyalinized and pink.
- This sometimes resulted in nodules of hyaline sclerosis separated by a background of loose myxoid material.
- The hyaline sclerotic material stained blue on Masson trichrome stains, indicating its collagenous nature.
Cellular pattern (26%)
- The cellular pattern of CCSK is characterized by less extracellular matrix material between cells with overlapping of nuclei, a feature that may lead to confusion with a blastemal predominant Wilms’ tumor or primitive neuroectodermal tumor. Mitotic activity is usually increased in this variant.
- The cellular pattern formed discrete, well-demarcated nodules within a less cellular background. In these areas, the intercellular material spacing the nuclei apart was diminished, and nuclear overlap was common.
- Mitotic activity in these areas often appeared increased, and the septa were limited to thin capillaries.
- The appearance approached that of an undifferentiated small, round, blue cell tumor, raising the differential diagnosis of PNET and blastemal Wilms tumor.
Epithelioid pattern (trabecular or acinar type) (13%)
- The epithelioid CCSK variant may be confused with nephroblastoma due to condensation of tumor cell cords.
- The epithelioid trabecular pattern featured plump cord cells uniformly retracted away from surrounding septa such that the cord cells aligned themselves into ribbons one or two cells thick.
- This appearance was highly reminiscent of the trabecular growth pattern of epithelial tumors such as hepatocellular carcinoma and thyroid follicular carcinoma.
- When viewed in cross section, the uniform alignment of these plump cord cells about the thin vascular septa created the appearance of a perivascular pseudorosette.
- When the central vessel was not visible, the appearance mimicked that of the Homer Wright rosettes of PNET or the true tubular differentiation of Wilms tumor.
- Like the cellular pattern, the trabecular pattern also tended to form discrete nodules within a tumor containing other CCSK patterns.
- An acinar epithelioid appearance occurred when the cord cells aligned their nuclei against the septa and their prominent eosinophilic cytoplasm toward the center of the cord cell clusters.
- In contrast to the true tubules of a Wilms tumor, such structures did not feature well-defined luminal borders.
Palisading (verocay-body) pattern (11%)
- The palisading pattern is described as having spindle cell nuclei in parallel linear arrays alternating with nuclear free zones, a feature that resembles Verocay bodies of schwannomas.
- The palisaded “verocay-body” pattern resulted from the alignment of spindled cord cells in parallel linear arrays with nuclei perpendicular to the septa.
- The septa were usually surrounded by collagenous material in these formations, creating a pink contrast to the aligned blue nuclei.
- The appearance raised the differential diagnosis of schwannoma, particularly in soft tissue metastasis.
Spindle cell pattern (7%)
- The spindle cell and storiform patterns are relatively uncommon.
- Spindle cell patterns resulted in a variety of appearances that suggested the diagnosis of other sarcomas.
- Spindled transformation of both cord and septal cells produced a storiform pattern of growth that mimicked fibrohistiocytic neoplasms.
- More commonly, the cord cell/septal cell demarcation was completely lost, resulting in a monomorphic high-grade sarcomatous appearance.
- In a spindled nucleus the fine chromatin characteristic of CCSK was more difficult to appreciate. In several cases, most of the sections showed a high-grade spindle cell sarcoma appearance, with the diagnosis of CCSK becoming evident only in small characteristic foci or in recurrences.
Storiform pattern (4%)
Anaplastic pattern (2.6%)
- Anaplasia is a rare finding in CCSK (3% of cases), and is characterized by the presence of enlarged, hyperchromatic polypoid nuclei with multipolar mitotic figures.
- The anaplastic pattern is defined by nuclear hyperchromasia, nuclear gigantism, and atypical mitoses.
- Overexpression of p53 (>75% of the nuclei) has been demonstrated in 2 of 3 anaplastic clear cell sarcoma of the kidney lesions.
- The nuclear accumulation of p53 in anaplastic tumors is thought to represent evidence of p53 gene mutation, a finding that has been well-documented in anaplastic Wilms’ tumors.
- Anaplasia, defined by nuclear hyperchromasia, nuclear gigantism, and atypical mitoses, was identified in rare tumors.
Nota bene: tumor cells in clear cell sarcoma of the kidney may appear misleadingly bland, particularly in the setting of metastatic disease following chemotherapy.
Immunohistochemistry
Immunohistochemistry is rarely informative in CCSK. Immunoreactivity for the intermediate filament vimentin is usually present, however, reactivity with most other proteins including epithelial markers are negative.
No diagnostic immunohistochemical stains allowing a definitive positive diagnosis of clear cell sarcoma (Argani et al 2000).
Vimentin and BCL2 are usually positive
Importantly, in the differential diagnosis with other pediatric renal malignancies:
- cytokeratin, WT1, CD99 staining is negative
- vimentin staining does not reveal the dot-like pattern of renal rhabdoid tumor.
Ultrastucture
Electron microscopy reveals features of primitive mesenchymal cells with abundant pale extracellular matrix, containing scant collagen fibers, and occasional septa, containing myofibroblasts or pericytes.
The main contribution of immunohistochemistry and electron microscopy is to exclude other diagnostic possibilities.
Differential diagnosis
CCSK may be confused with Wilms’ tumor, mesoblastic nephroma, and malignant rhabdoid tumor of the kidney.
Diagnosis is a morphological one, in part a diagnosis of exclusion. On the basis of resection specimens, numerous areas of the tumor may be examined and focally characteristic and diagnostic areas may be seen.
Needle biopsy interpretation may be difficult since the varying patterns of clear cell sarcoma of the kidney may mimic a variety of other tumors.
Staging
Stage 1: Tumor confined to the kidney and completely resected. No penetration of the renal capsule or involvement of renal sinus vessels.
Stage 2: Tumor extends beyond kidney but completely resected. Tumor penetrates renal capsule, invades sinus vessels, was biopsied before removal, or spilled locally during removal, but margins are negative.
Stage 3: Gross residual tumor, positive surgical margins, massive tumor spill or lymph node metastases.
Stage 4: Hematogenous metastases.
Stage 5: Bilateral renal tumors.
The revised tumor stage distribution at presentation for NWTS, using NWTS 5 criteria, was as follows:
25% of patients had localized stage 1 tumors,
a majority of patients presented with stage 2 (37%) or 3 (34%) disease,
only 4% of patients presented with distant metastasis (stage 4).
No true bilateral primary tumors were identified.
In one case of the NWTS study, a 1-cm tumor was found in the kidney contralateral to a kidney harboring a 13-cm CCSK. Because the patient presented with widely disseminated stage IV disease, this the contralateral tumor has been interpreted as a metastasis.
No one histologic pattern is overrepresented in lymph node metastases at presentation. Occasionally, a pattern not seen in the primary tumor is evident in its metastasis. However, the entrapped renal tubules and the epithelioid patterns seen in primary tumors are not encountered in a metastasis.
Post-treatment relapses generally displayed the same range of patterns seen in primary tumors, with the notable absence of the epitheliod pattern. However, a trend toward more sclerotic, less cellular lesions was seen, possibly reflecting treatment effect.
These non-pleomorphic, sclerotic relapses often entrapped native epithelial structures (bile ducts, alveoli) in their metastatic sites, mimicking benign bile duct or pulmonary hamartomas. Two unique patterns are identified among the metastases.
The first is a hypocellular spindle cell pattern that simulated fibromatosis. On diligent search, small residual foci of epithelioid cord cells could be identified, allowing a diagnosis of metastatic CCSK to be made with confidence.
The second is a paucicellular myxoid pattern that featured pools of myxoid material bearing only rare free-floating tumor cells. 9 Confusion with myxoma was heightened by a focal absence of the characteristic CCSK vasculature.
Cytogenetics
t(10;17)(q22;p13) (15474157, 2548705, 17516747)
del(14)(q24.1q31.1) (17516747)
Only a small number of CCSK cases have been described cytogenetically. A clonal reciprocal 10;17 translocation t(10;17)(q22;p13) in CCSK was first reported in 1989.
A CCSK with a complex karyotype including trisomy 9, deletions of chromosomes 16 and 22, and loss of chromosome 1p13 has been reported.
In the same case, an interstitial deletion of chromosome 14 was reported: del(14)(q23).
One of two "sarcomatous Wilms’ tumors" also contained a t(10;17)(q11;p12) as a part of an abnormal karyotype. Three of four other patients with CCSK were normal whereas one patient harbored a t(2;22)(q21;q11).
Of five patients reviewed at this institution, karyotypes were available for four of these. One patient had a clonal balanced translocation 10;17 and an interstitial deletion of the long arm of chromosome 14 as follows: 46, XY, t(10;17)(q22;p13)del(14)(q24.1q31.1).Three other patients had normal karyotypes. Fluorescent in-situ hybridization using a p53 probe was employed on the same cells harboring the clonal translocation above. This study documented the presence of two p53 signals on chromosome 17 indicating the absence of deletion or translocation of the TP53 tumor suppressor gene.
CGH (10657872)
Comparative genomic hybridization analysis of CCSK has documented quantitative chromosomal abnormalities in only 4 of 30 CCSK cases.
These four cases included a gain of chromosome 1q and loss of 10q, gain of 1q and loss of terminal 4p, gain of 19p, and loss of chromosome 19. Later, another CCSK with a t(10;17)(q22;p13) was reported.
Molecular biology
LOI for IGF2 (43%) (13679437)
Treatment
Treatment of CCSK generally involves surgical intervention coupled with radiation and chemotherapy. CCSK commonly responds poorly to treatment with vincristine and actinomycin alone, but the addition of doxorubicin to chemotherapy regimens has improved survival rates.
In the NWTS-5 protocol, patients with all stages of CCSK are treated with the same regimen used in patients who have Wilms tumor with diffuse anaplasia with the exception of stage I tumors.
This treatment protocol is comprised of radical nephrectomy followed by radiotherapy and chemotherapy with cyclophosphamide, etoposide, vincristine, and doxorubicin for 24 weeks.
Prognosis
Stage I (25% of CCSK): six-year survival is 97%
Stage II (37% of CCSK): 75%
Stage III (34% of CCSK): 77%
Stage IV (4% of CCSK): 50%
The prognosis for CCSK, particularly for low stage tumors, has improved with the addition of doxorubicin to chemotherapy regimens with a 66% reduction in overall mortality.
Stage-dependent six-year survival is 97% for stage I tumors, 75% for stage II tumors, 77% for stage III tumors, and 50% for stage IV tumors.
Patients with tumors without areas of necrosis have a more favorable prognosis. Twenty-nine percent of patients with CCSK have lymph node metastases at the time of diagnosis, and bone metastasis is the most common form of relapse.
Metastatic lesions have also been reported in the liver, brain, soft tissue sites, and lung with more unusual metastases to the skeletal muscle, testis, and salivary gland. Relapses of CCSK as many as 10 years after original diagnosis have been reported.
Molecualr biology
BCOR internal tandem duplications (ITD) in :
- Ewing-like sarcoma (27000436)
- clear cell sarcoma of the kidney (CCSK) (26098867, 26874462, 27000436, 26573325)
Open references
Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Roy A, Kumar V, Zorman B, Fang E, Haines KM, Doddapaneni H, Hampton OA, White S, Bavle AA, Patel NR, Eldin KW, John Hicks M, Rakheja D, Leavey PJ, Skapek SX, Amatruda JF, Nuchtern JG, Chintagumpala MM, Wheeler DA, Plon SE, Sumazin P, Parsons DW.
Nat Commun. 2015 Nov 17;6:8891. doi : 10.1038/ncomms9891
PMID: 26573325 Free
Paywall References
Recurrent BCOR Internal Tandem Duplication and YWHAE-NUTM2B Fusions in Soft Tissue Undifferentiated Round Cell Sarcoma of Infancy: Overlapping Genetic Features With Clear Cell Sarcoma of Kidney.
Kao YC, Sung YS, Zhang L, Huang SC, Argani P, Chung CT, Graf NS, Wright DC, Kellie SJ, Agaram NP, Ludwig K, Zin A, Alaggio R, Antonescu CR.
Am J Surg Pathol. 2016 Aug;40(8):1009-20. doi : 10.1097/PAS.0000000000000629
PMID: 26945340
BCOR Overexpression Is a Highly Sensitive Marker in Round Cell Sarcomas With BCOR Genetic Abnormalities.
Kao YC, Sung YS, Zhang L, Jungbluth AA, Huang SC, Argani P, Agaram NP, Zin A, Alaggio R, Antonescu CR.
Am J Surg Pathol. 2016 Jul 13.
PMID: 27428733
Mutually exclusive BCOR internal tandem duplications and YWHAE-NUTM2 fusions in clear cell sarcoma of kidney: not the full story. Kenny C, Bausenwein S, Lazaro A, Furtwängler R, Gooskens SL, van den Heuvel Eibrink M, Vokuhl C, Leuschner I, Graf N, Gessler M, O’Sullivan MJ.
J Pathol. 2016 Apr;238(5):617-20. doi : 10.1002/path.4693
PMID: 27000436
BCOR internal tandem duplications in clear cell sarcoma of the kidney.
Gooskens SL, Gadd S, van den Heuvel-Eibrink MM, Perlman EJ.
Genes Chromosomes Cancer. 2016 Jun;55(6):549-50. doi : 10.1002/gcc.22353. No abstract available
PMID: 26874462
Consistent in-frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney.
Ueno-Yokohata H, Okita H, Nakasato K, Akimoto S, Hata J, Koshinaga T, Fukuzawa M, Kiyokawa N.
Nat Genet. 2015 Aug;47(8):861-3. doi : 10.1038/ng.3338
PMID: 26098867
Links
Reference articles
Clear cell sarcoma of the kidney: a review of 351 cases from the National Wilms Tumor Study Group Pathology Center. Argani P, Perlman EJ, Breslow NE, Browning NG, Green DM, D’Angio GJ, Beckwith JB. Am J Surg Pathol. 2000 Jan;24(1):4-18. PMID: 10632483
References - Cytogenetics
Recurring translocation (10;17) and deletion (14q) in clear cell sarcoma of the kidney. Brownlee NA, Perkins LA, Stewart W, Jackle B, Pettenati MJ, Koty PP, Iskandar SS, Garvin AJ. Arch Pathol Lab Med. 2007 Mar;131(3):446-51. PMID: 17516747
Translocation (10;17)(q22;p13): a recurring translocation in clear cell sarcoma of kidney. Rakheja D, Weinberg AG, Tomlinson GE, Partridge K, Schneider NR. Cancer Genet Cytogenet. 2004 Oct 15;154(2):175-9. PMID: 15474157
Translocation (10;17)(q22;p13): a recurring translocation in clear cell sarcoma of kidney. Rakheja D, Weinberg AG, Tomlinson GE, Partridge K, Schneider NR. Cancer genetics and cytogenetics. 2004 ; 154 (2) : 175-179. PMID 15474157
Genetic and genetic expression analyses of clear cell sarcoma of the kidney. Schuster AE, Schneider DT, Fritsch MK, Grundy P, Perlman EJ. Lab Invest. 2003 Sep;83(9):1293-9. PMID: 13679437
Comparative genomic hybridization analysis of clear cell sarcoma of the kidney. Barnard M, Bayani J, Grant R, Zielenska M, Squire J, Thorner P. Med Pediatr Oncol. 2000 Feb;34(2):113-6. PMID: 10657872
Clear cell sarcoma of kidney in an adolescent and in young adults: a report of four cases with ultrastructural, immunohistochemical, and DNA flow cytometric analysis. Amin MB, de Peralta-Venturina MN, Ro JY, El-Naggar A, Mackay B, Ordonez N, Mani A, Ayala A. Am J Surg Pathol. 1999 Dec;23(12):1455-63. PMID: 10584698
Translocation 10;17 in clear cell sarcoma of the kidney. A first report. Punnett HH, Halligan GE, Zaeri N, Karmazin N. Cancer Genet Cytogenet. 1989 Aug;41(1):123-8. PMID: 2548705
Abnormalities of chromosomes 1 and 11 in Wilms’ tumor.
Douglass EC, Wilimas JA, Green AA, Look AT. Cancer genetics and cytogenetics. 1985 ; 14 (3-4) : 331-338. PMID 2981607
Translocation 10;17 in clear cell sarcoma of the kidney. A first report. Punnett HH, Halligan GE, Zaeri N, Karmazin N. Cancer genetics and cytogenetics. 1989 ; 41 (1) : 123-128. PMID #2548705"
Chromosome analysis of 31 Wilms’ tumors. Sheng WW, Soukup S, Bove K, Gotwals B, Lampkin B. Cancer research. 1990 ; 50 (9) : 2786-2793. PMID 2158398
Comparative genomic hybridization analysis of clear cell sarcoma of the kidney. Barnard M, Bayani J, Grant R, Zielenska M, Squire J, Thorner P. Medical and pediatric oncology. 2000 ; 34 (2) : 113-116. PMID 10657872
Functional and gene expression analysis of the p53 signaling pathway in clear cell sarcoma of the kidney and congenital mesoblastic nephroma. Brownlee NA, Hazen-Martin DJ, Garvin AJ, Re GG. Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society. 2002 ; 5 (3) : 257-268. PMID 12007018
Infrequent p53 gene mutations and lack of p53 protein expression in clear cell sarcoma of the kidney: immunohistochemical study and mutation analysis of p53 in renal tumors of unfavorable prognosis. Hsueh C, Wang H, Gonzalez-Crussi F, Lin JN, Hung IJ, Yang CP, Jiang TH. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2002 ; 15 (6) : 606-610. PMID 12065773
References - Morphology
Clear cell sarcoma of the kidney: a review of 351 cases from the National Wilms Tumor Study Group Pathology Center. Argani P, Perlman EJ, Breslow NE, Browning NG, Green DM, D’Angio GJ, Beckwith JB. Am J Surg Pathol. 2000 Jan;24(1):4-18. PMID: 10632483
Correlation of chromosome abnormalities with histological and clinical features in Wilms’ and other childhood renal tumors. Kaneko Y, Homma C, Maseki N, Sakurai M, Hata J. Cancer research. 1991 ; 51 (21) : 5937-5942. PMID 1657374
Clear cell sarcoma of kidney. Two cases in adults. Oda H, Shiga J, Machinami R. Cancer. 1993 ; 71 (7) : 2286-2291. PMID 8453549
Implications of p53 protein expression in clear cell sarcoma of the kidney.
Cheah PL, Looi LM. Pathology. 1996 ; 28 (3) : 229-231. PMID 8912350
Clear cell sarcoma of the kidney: a review of 351 cases from the National Wilms Tumor Study Group Pathology Center. Argani P, Perlman EJ, Breslow NE, Browning NG, Green DM, D’Angio GJ, Beckwith JB. The American journal of surgical pathology. 2000 ; 24 (1) : 4-18. PMID 10632483
Beckwith JB. Renal tumors. In: Stocker JT, Askin FB, eds. Pathology of Solid Tumors in Children. New York, NY: Chapman & Hall Medical, 1998: 1–23.
Kodet R, Stejskal J, Malis J, Horak J. Bone metastasizing renal tumor of childhood. A clinicopathological study of eleven cases from the Prague Pediatric Tumor Registry. Pathol Res Pract 1994; 190:750–8.
Green DM, Breslow NE, Beckwith JB, Moksness J, Finklestein JZ, D’Angio GJ. Treatment of children with clear-cell sarcoma of the kidney: a report from the National Wilms’ Tumor Study Group. J Clin Oncol 1994; 12:2132–7.
Looi LM, Cheah PL. An immunohistochemical study comparing clear cell sarcoma of the kidney and Wilms’ tumor. Pathology 1993; 25:106–9.
Feusner JH, Beckwith JB, D’Angio GJ. Clear cell sarcoma of the kidney: accuracy of imaging methods for detecting bone metastases. Report from the National Wilms’ Tumor Study. Med Pediatr Oncol 1990; 18:225–7.
Florine BL, Simonton SC, Sane SM, Stickel FR, Singher LJ, Dehner LP. Clear cell sarcoma of the kidney: report of a case with mandibular metastasis simulating a benign myxomatous tumor. Oral Surg Oral Med Oral Pathol 1988; 65:567–74.
Ogawa K, Nakashima Y, Yamabe H, Hamashima Y. Clear cell sarcoma of the kidney. An immunohistochemical study. Acta Pathologica Japanica 1986; 36:681–9.
Schmidt D, Harms D, Evers KG, Bliesener JA, Beckwith JB. Bone metastasizing renal tumor (clear cell sarcoma) of childhood with epithelioid elements. Cancer 1985; 56:609–13.
Knisely AS, Magid MS, Alonso D, Hilgartner M. Clear-cell sarcoma of the kidney can coexpress cytokeratin and vimentin [Abstract]. Lab Invest 1985; 56:4P.
Sotelo-Avila C, Gonzalez-Crussi F, Sadowinski S, Gooch WM, Pena R. Clear cell sarcoma of the kidney: a clinicopathologic study of 21 patients with long-term follow-up evaluation. Hum Pathol 1985; 16:1219–30.
Haas JE, Bonadio JF, Beckwith JB. Clear cell sarcoma of the kidney with emphasis on ultrastructural studies. Cancer 1984; 54:2978–87.
Undifferentiated sarcoma of the kidney: a tumor of childhood with histopathologic and clinical characteristics distinct from Wilms’ tumor.
Morgan E, Kidd JM. Cancer. 1978 ; 42 (4) : 1916-1921. PMID 213187
Bone-metastasizing renal tumour of childhood. Marsden HB, Lawler W. British journal of cancer. 1978 ; 38 (3) : 437-441. PMID 708576
Marsden HB, Lawler W. Bone-metastasizing renal tumour of childhood. Br J Cancer 1978; 38:437–41.
Marsden HB, Lawler W. Bone metastasizing renal tumour of childhood. Histopathological and clinical review of 38 cases. Virchows Arch [Pathol Anat] 1980; 387:341–51.
Marsden HB, Lawler W, Kumar PM. Bone metastasizing renal tumor of childhood: morphological and clinical features, and differences from Wilms’ tumor. Cancer 1978; 42:1922–8.
Morgan E, Kidd JM. Undifferentiated sarcoma of the kidney: a tumor of childhood with histopathologic and clinical characteristics distinct from Wilms’ tumor. Cancer 1978; 42:1916–21.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}