Home > D. General pathology > Genetic and developmental anomalies > Genetic metabolic diseases > glycogen storage disease type 4
glycogen storage disease type 4
MIM.232500
Sunday 25 April 2004
Digital cases
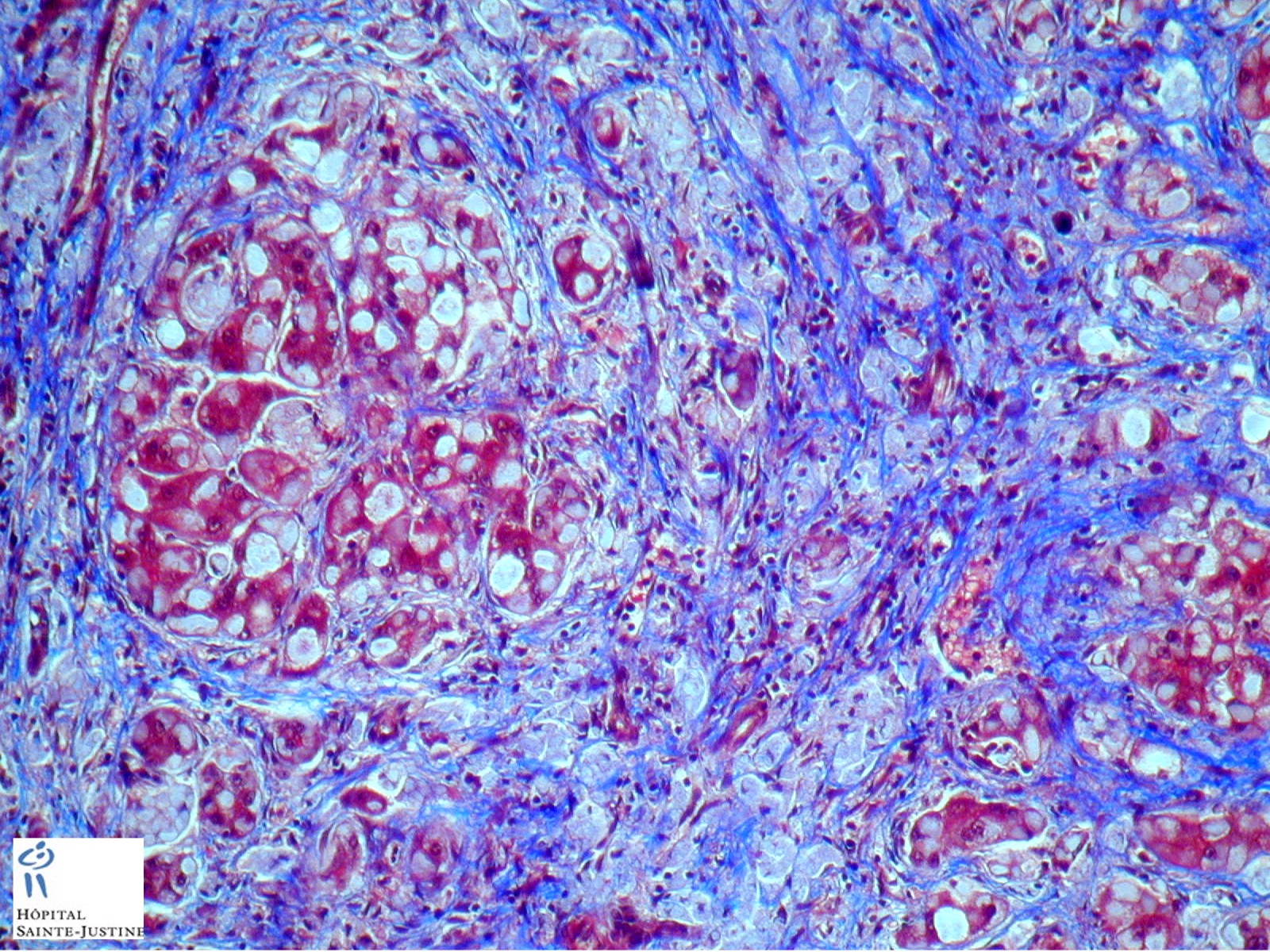
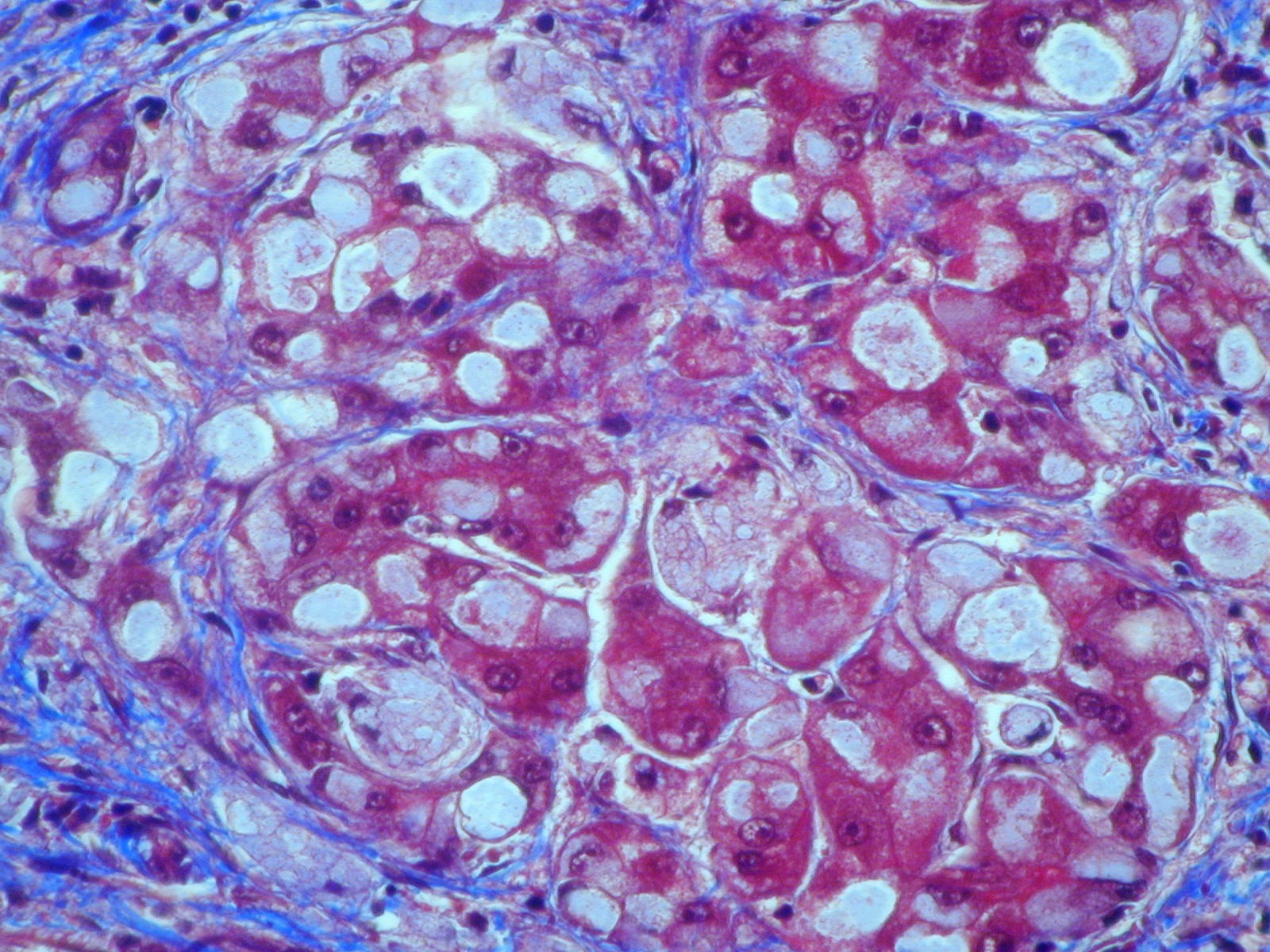
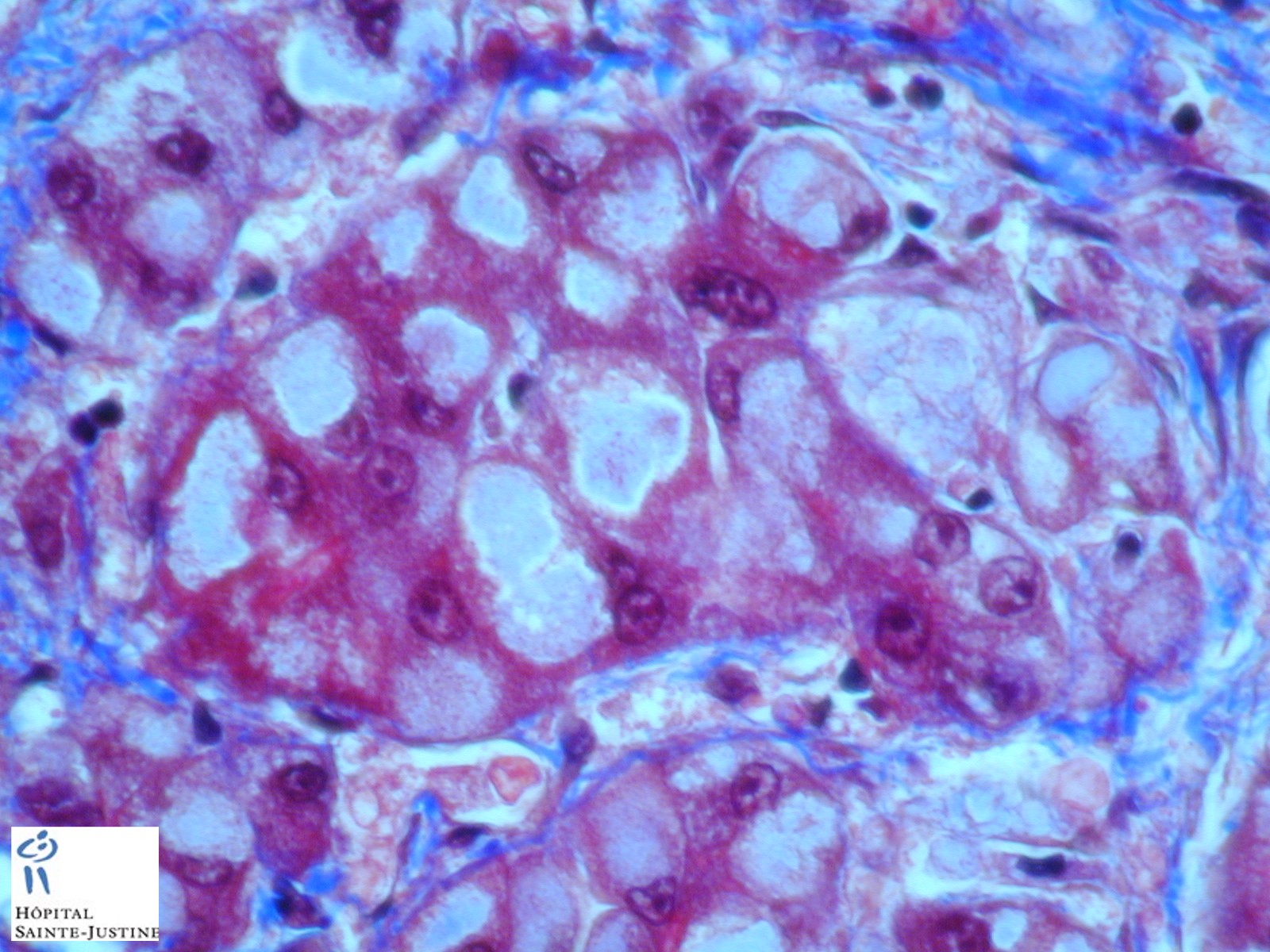
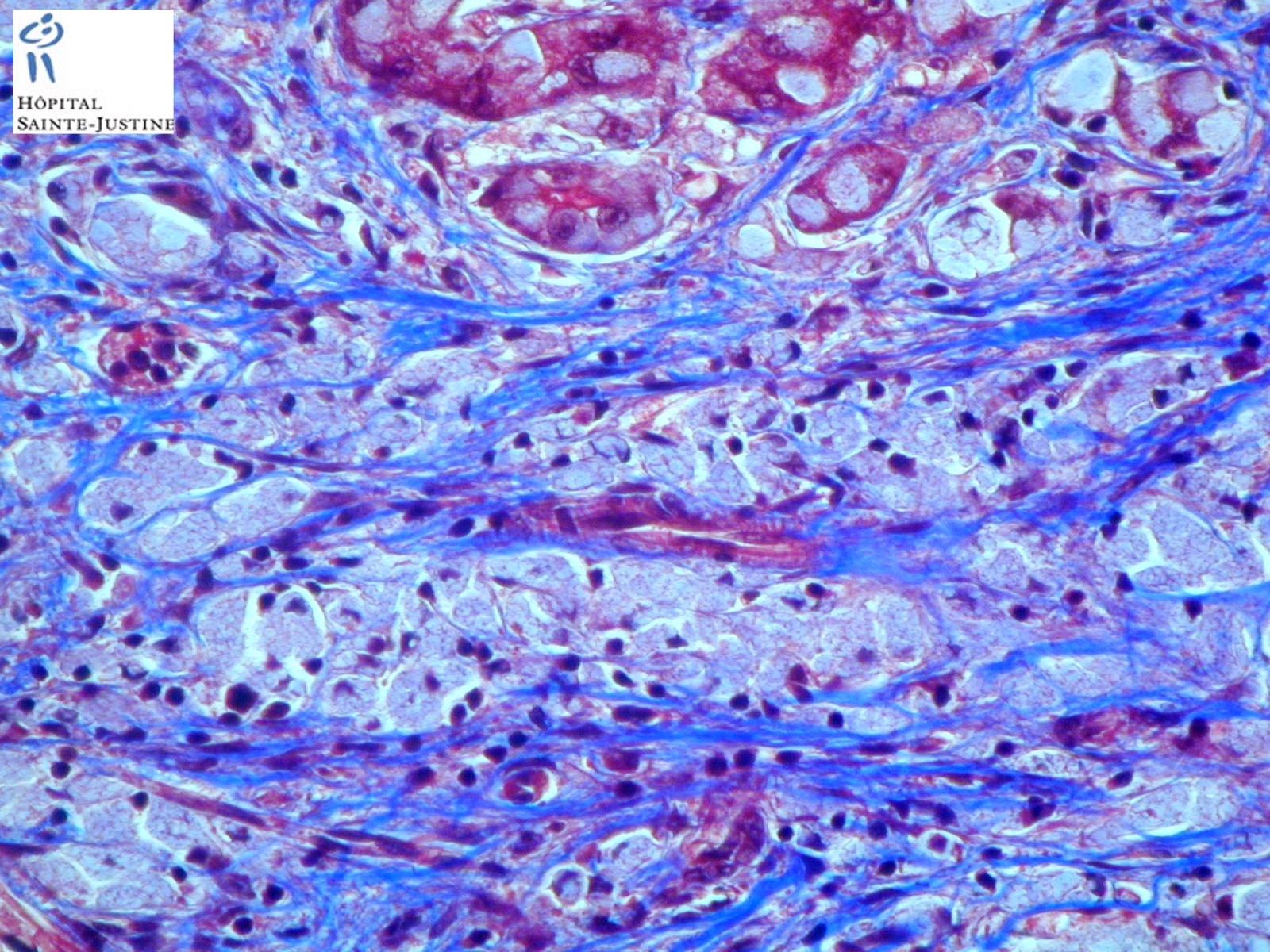
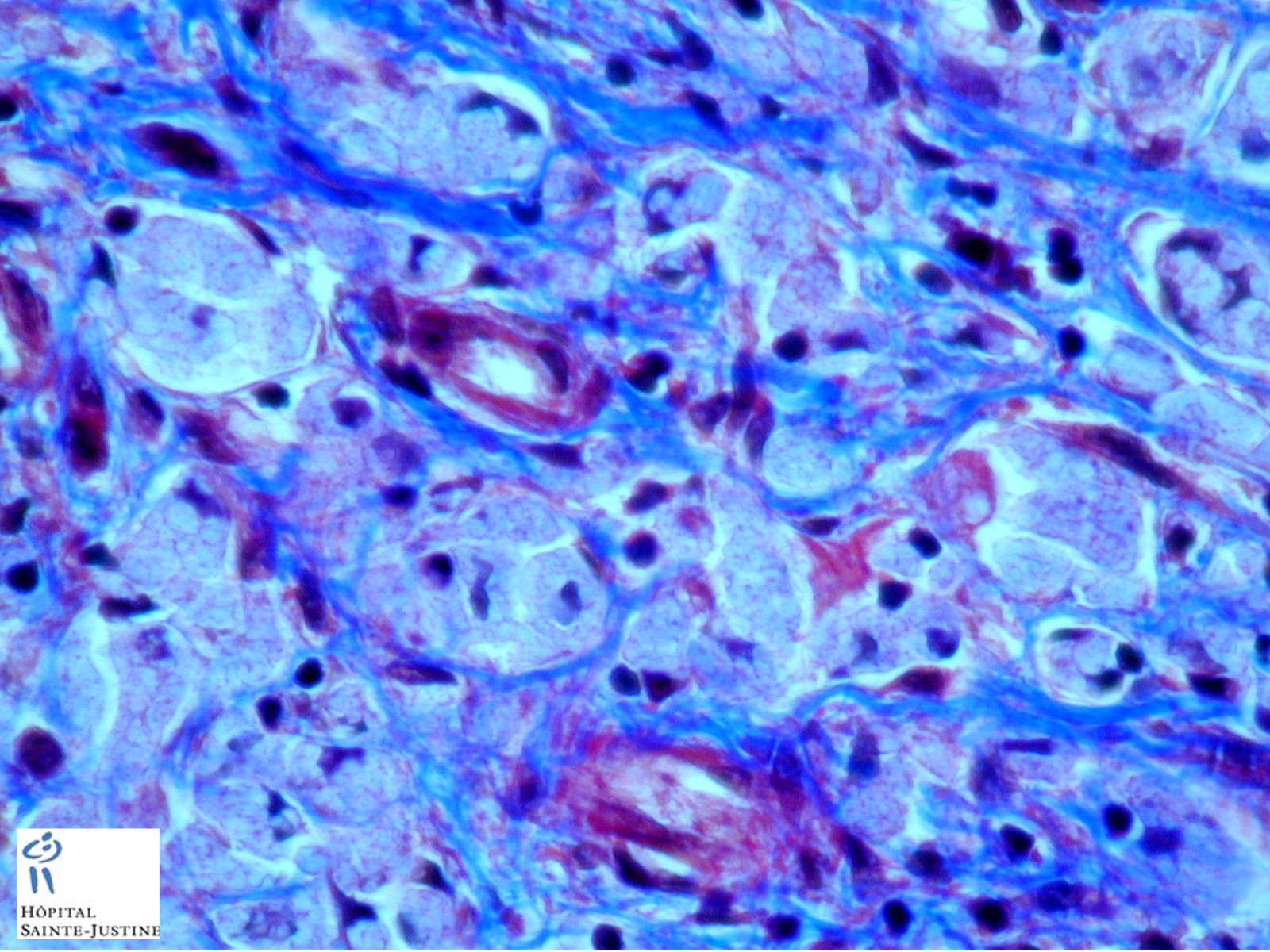
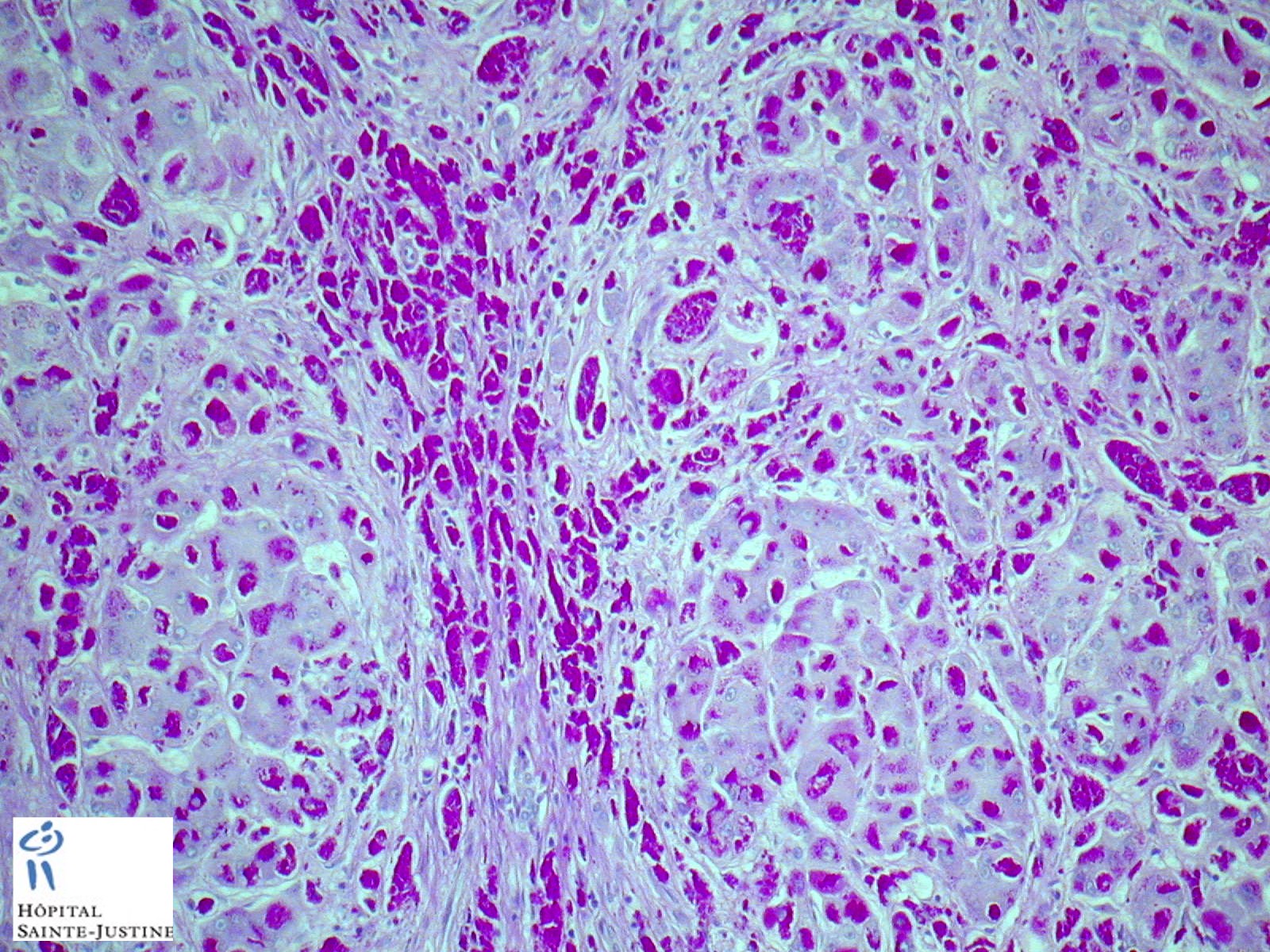
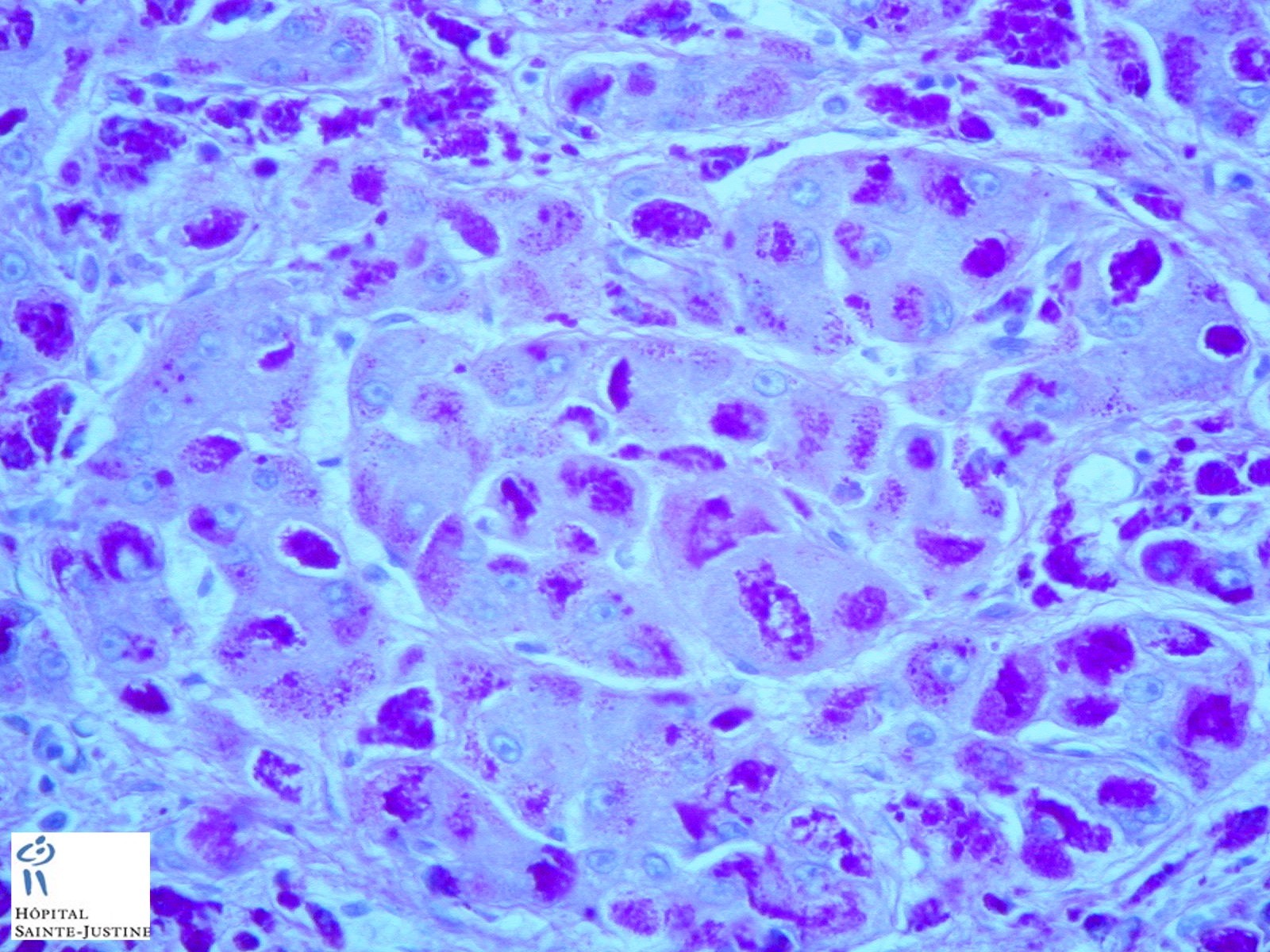
HPC:403 : Cirrhosis in glycogen storage disease type 4 (GSD4)
Definition: Glycogen storage disease type IV (GSD IV, or Andersen disease) is an autosomal recessive disorder due to the deficiency of 1,4-alpha-glucan branching enzyme (or glycogen branching enzyme, GBE1), resulting in an accumulation of amylopectin-like polysaccharide in muscle, liver, heart and central and peripheral nervous system.
Clinical synopsis
Clinical findings of branching enzyme deficiency relate to liver disease and include hepatic failure, cirrhosis, hepatosplenomegaly, cardiomyopathy (less frequent), failure to thrive, and hypotonia (in some cases). Ventricular arrhythmia may occur.
Typically, the presentation is in childhood with liver involvement up to cirrhosis. The neuromuscular form varies in onset (congenital, perinatal, juvenile and adult) and in severity.
Congenital cases are rare, and fewer than 20 cases have been described and genetically determined so far. This form is characterized by polyhydramnios, neonatal hypotonia, and neuronal involvement; hepatopathy is uncommon, and the babies usually die between 4 weeks and 4 months of age.
Synopsis
digestive anomalies
- numerous large macrophages in the mucosa
Some inclusions were PAS positive and others were not.
Two kinds of polysaccharides can be noted in the cardiac muscle, skeletal muscles, smooth muscles and reticuloendothelial cells, but also in the neutrophils and platelets. One was glycogen and the other was similar to amylopectin.
- hepatic fibrosis
- periodic-acid Schiff-positive, diastase-resistant inclusions
- fibrillar inclusions characteristic of amylopectin by electron microscopy.
Nota bene: Liver biopsy may be needed to determine the cause of progressive liver dysfunction. Histologic findings are characteristic in the liver, with diffuse interstitial fibrosis, wide fibrous septa, and enlarged hepatocytes with periodic acid-Schiff positive inclusions. Electron microscopy shows alpha and beta glycogen particles.
Physiopathology
Glycogen storage disease type IV is a result of the absence of the glycogen branching enzyme amylo-1,4-1,6 transglucosidase, which is critical in the production of glycogen. This leads to very long unbranched glucose chains, forming amylopectin-like polysaccharide.
The long unbranched molecules (known as amylopectin-like polysaccharide - see also amylopectin) have a low solubility which leads to glycogen precipitation in the liver. These deposits subsequently build up in the body tissue, especially the heart and liver. The probable end result is cirrhosis and death by age 5 years.
Transglucosidase, which is found in all tissues, is deficient. The condition is autosomal recessive. Due to abnormal glycogen, hepatic deposition may occur and result in severe cirrhosis, hepatic failure, or neuromuscular failure. It also can present as abnormal liver function tests in its mildest presentation.
Cardiac and skeletal muscle may show PAS+ eosinophilic cytoplasmic inclusions.
Biochemistry
Branching glycosyltransferase deficiency can be biochemically assessed.
Enzymatic assay reveals deficient hepatic branching enzyme activity with normal activity of glucose-6-phosphatase, debranching enzyme and phosphorylase activities.
Etiology
germline mutation in GBE1, coding for the glycogen branching enzyme (MIM.607839)
- mutation in the GBE1 gene also causes an allelic disorder, adult polyglucosan body disease (APED) (MIM.263570)
- GBE1 biochemical activity can be virtually absent in muscle and fibroblasts, and totally lacking in liver and heart as well as glycogen synthase activity.
- Truncating GBE1 mutations cause a spectrum of severe diseases ranging from generalized intrauterine hydrops to fatal perinatal hypotonia and fatal cardiomyopathy in the first months of life.
Variants
fatal perinatal GSD4
fetal GSD4
congenital neuromuscular GSD4
childhood neuromuscular GSD4
nonprogressive hepatic GSD4
classic hepatic GSD4
combined hepatic and muscular GSD4
adult polyglucosan body disease (APBD) (MIM.263570)
Ultrastructure (18661138)
pleomorphic polyglucosan bodies in muscle fibers and macrophages, and less severe in Schwann cells and microglial cells.
inclusions are granular and membrane-bound.
others had an irregular contour, are more electron dense and are not membrane bound, or homogenous (’hyaline’).
A paracrystalline pattern of granules is repeatedly noted showing a periodicity of about 10 nm with an angle of about 60 degrees or 120 degrees at sites of changing linear orientation.
Malteser crosses are noted under polarized light in the larger inclusions.
Large amount of fibrils, 60 A in width, glycogen rosettes and glycogen granules can be detected in overload cells.
See also
glycogen storage diseases
- amylopectinosis (amylopectinoses)
Links
References
Congenital type IV glycogenosis: the spectrum of pleomorphic polyglucosan bodies in muscle, nerve, and spinal cord with two novel mutations in the GBE1 gene. Nolte KW, Janecke AR, Vorgerd M, Weis J, Schröder JM. Acta Neuropathol. 2008 Nov;116(5):491-506. PMID: 18661138
Non-lethal neonatal neuromuscular variant of glycogenosis type IV with novel GBE1 mutations. Fernandez C, Halbert C, De Paula AM, Lacroze V, Froissart R, Figarella-Branger D, Chabrol B, Pellissier JF. Muscle Nerve. 2009 Oct 7. PMID: 19813197
Neuropathological study of skeletal muscle, heart, liver, and brain in a neonatal form of glycogen storage disease type IV associated with a new mutation in GBE1 gene. Lamperti C, Salani S, Lucchiari S, Bordoni A, Ripolone M, Fagiolari G, Fruguglietti ME, Crugnola V, Colombo C, Cappellini A, Prelle A, Bresolin N, Comi GP, Moggio M. J Inherit Metab Dis. 2009 Apr 8. PMID: 19357989
Bruno C, van Diggelen OP, Cassandrini D, Gimpelev M, Giuffre B, Donati MA, Introvini P, Alegria A, Assereto S, Morandi L, Mora M, Tonoli E, Mascelli S, Traverso M, Pasquini E, Bado M, Vilarinho L, van Noort G, Mosca F, DiMauro S, Zara F, Minetti C. Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV). Neurology. 2004 Sep 28;63(6):1053-8. PMID: 15452297
van Noort G, Straks W, Van Diggelen OP, Hennekam RC. A congenital variant of glycogenosis type IV. Pediatr Pathol. 1993 Sep-Oct;13(5):685-98. PMID: 8247964
Penchansky L, Agostini RM, Jaffe R. Leukocyte inclusions in glycogen storage disease, type IV. Pediatr Pathol. 1992 Nov-Dec;12(6):903-5. PMID: 1333075
A new variant of type IV glycogenosis: deficiency of branching enzyme activity without apparent progressive liver disease. Greene HL, Brown BI, McClenathan DT, Agostini RM Jr, Taylor SR. Hepatology. 1988 Mar-Apr;8(2):302-6. PMID: 3162725
Type IV glycogenosis - a study of two cases. Ishihara T, Uchino F, Adachi H, Takahashi M, Watanabe S, Tsunetoshi S, Fuji T, Ikee Y. Acta Pathol Jpn. 1975 Sep;25(5):613-33. PMID: 1060362
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}