Home > A. Molecular pathology > SOD1
SOD1
Tuesday 23 September 2003
Locus: 21q22.1
Pathology
Mutations in the SOD1 gene are the most common form of inherited ALS, accounting for 20% of all the familial ALS forms and corresponding to 1%–2% of all ALS cases.
Since the first SOD1 missense mutations were reported in 1993, the number of known mutations has increased to more than 114. These are distributed throughout all five exons encoding the 153 amino acid protein, with no region of the polypeptide escaping disease causing mutation. With the exception of a few instances, all SOD1 mutations are dominant.
Sporadic and SOD1 mutant-mediated familial ALS are clinically indistinguishable and affect the same neurons, but even within the familial forms, the time of onset and length of disease vary.
The alanine-to-valine substitution at position 4 of SOD1 (SOD1A4V) is the most prominent mutation in North America, responsible for 50% of cases, and has a uniformly aggressive disease course with a mean survival of only 1 year after onset.
Mice and rats expressing mutant forms of human or mouse SOD1 develop progressive motor neuron degeneration.
Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis.
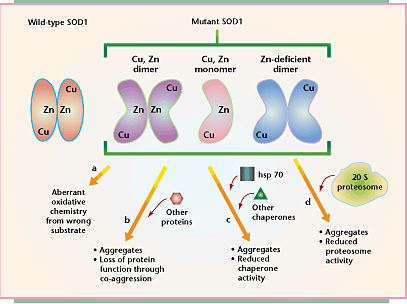
Several models have been proposed for SOD1-mediated toxicity linked to altered conformation and/or aggregation of mutant SOD1 subunits. Wild-type SOD1 exists as a dimer with bound Cu and Zn, while mutants are misfolded, unable to dimerize or unable to efficiently bind Zn.
Mutant SOD1 toxicity may result from aberrant oxidative chemistry. Misfolded enzymatic subunits admit access of improper substrates to the catalytic copper site of SOD1.
Neuronal toxicity arises from protein aggregation. Diffuse or focal aggregates sequester key cellular components.
Aggregates place an increased burden on protein chaperones, which continually try to fold or refold mutant SOD1, leading to a reduction in overall protein-folding chaperone activity.
Accumulation of mutant SOD1 overwhelms the capacity of the ubiquitin proteasome pathway to degrade malfolded SOD molecules, but also other critical substrates.
The instability of the mutant protein contributes to its toxicity, sometimes enhanced by the release of Zn.
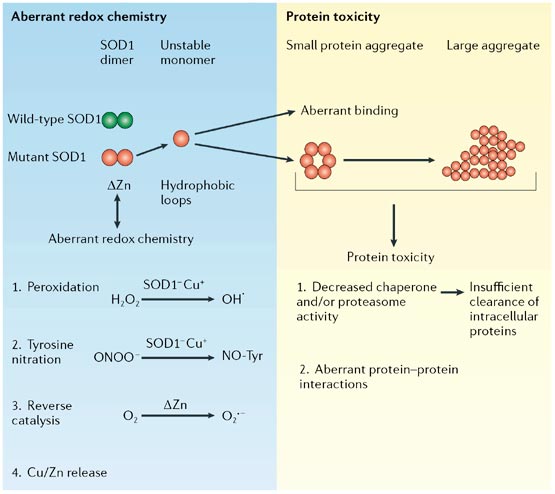
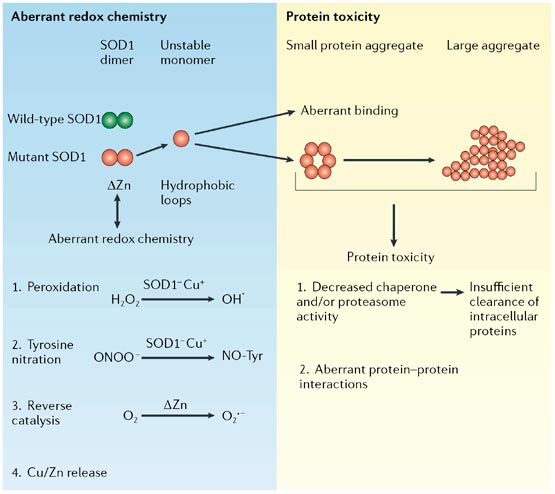
In the aberrant redox chemistry model, mutant superoxide dismutase 1 (SOD1) is unstable and the active channel opens, permitting aberrant chemistry through promiscuous interaction with non-conventional substrates.
Hydrogen peroxide (H2O2) or nitronium ion (ONOO-) can react with reduced SOD1 (SOD1-Cu+). Molecular oxygen (O2) can react aberrantly with Zn-deficient SOD1 to generate an excess of superoxide anion (O2-). The unstable protein could also release free copper and/or zinc, which might be toxic.
In the protein toxicity model, unstable, conformationally altered mutant SOD1 could form toxic, proteinaceous deposits.
Aggregated SOD1 could inhibit chaperone and/or proteasome activity, with subsequent misfolding and insufficient clearance of numerous proteins.
Alternatively, these aggregates could sequester, inactivate or enhance the toxicity of other proteins crucial for cellular processes.
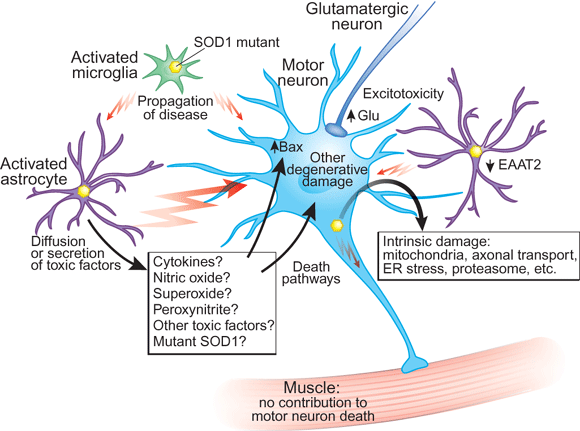
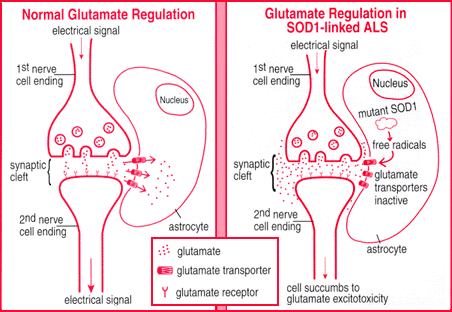
Mutant SOD1 caused intrinsic damage to motor neurons. The reduction of mutant SOD1 in motor neurons delays the onset of disease and extends the lifespan of transgenic ALS mice.
Reduction of mutant SOD1 in microglia affects disease duration after onset, suggesting a role for microglia in the propagation of the disease. Although there is cell-autonomous damage caused by mutant SOD1 in the motor neurons, this may not be sufficient to trigger and propagate ALS pathogenesis.
In vitro experiments suggest that activated astrocytes might be important mediators of mutant SOD1 toxicity that results in motor neuron death.
Physiopathology
The instability of the mutant protein contributes to its toxicity, sometimes enhanced by the release of Zn. In the aberrant redox chemistry model, mutant superoxide dismutase 1 (SOD1) is unstable and the active channel opens, permitting aberrant chemistry through promiscuous interaction with non-conventional substrates. Hydrogen peroxide (H2O2) or nitronium ion (ONOO-) can react with reduced SOD1 (SOD1-Cu+).
Molecular oxygen (O2) can react aberrantly with Zn-deficient SOD1 to generate an excess of superoxide anion (O2-). The unstable protein could also release free copper and/or zinc, which might be toxic. In the protein toxicity model, unstable, conformationally altered mutant SOD1 could form toxic, proteinaceous deposits.
Aggregated SOD1 could inhibit chaperone and/or proteasome activity, with subsequent misfolding and insufficient clearance of numerous proteins. Alternatively, these aggregates could sequester, inactivate or enhance the toxicity of other proteins crucial for cellular processes.
See also
ALS genes
| SOD1 | ALS2 | SETX | VAPB | ANG | DCTN1 | MAPT |
References
Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006 Sep;7(9):710-23. PMID: 16924260
Jean-Pierre Julien. ALS: astrocytes move in as deadly neighbors. Nature Neuroscience 10, 535 - 537 (2007)
Bendotti C, Carri MT. Lessons from models of SOD1-linked familial ALS. Trends Mol Med. 2004 Aug;10(8):393-400. PMID: 15310460
Don W. Cleveland & Jian Liu. Oxidation versus aggregation — how do SOD1 mutants cause ALS? Nature Medicine 6, 1320 - 1321 (2000).
Don W. Cleveland1 & Jeffrey D. Rothstein. From charcot to lou gehrig: deciphering selective motor neuron death in als. Nature Reviews Neuroscience 2, 806-819 (November 2001)
MIM.147450
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}