Home > D. General pathology > Blood and immunity > Griscelli disease
Griscelli disease
Wednesday 17 September 2003
Definition: Griscelli syndrome (GS), a rare autosomal recessive disorder, is characterized by partial albinism, along with immunologic abnormalities or severe neurological impairment or both. Mutations in one of two different genes on chromosome 15q can cause the two different subtypes of GS.
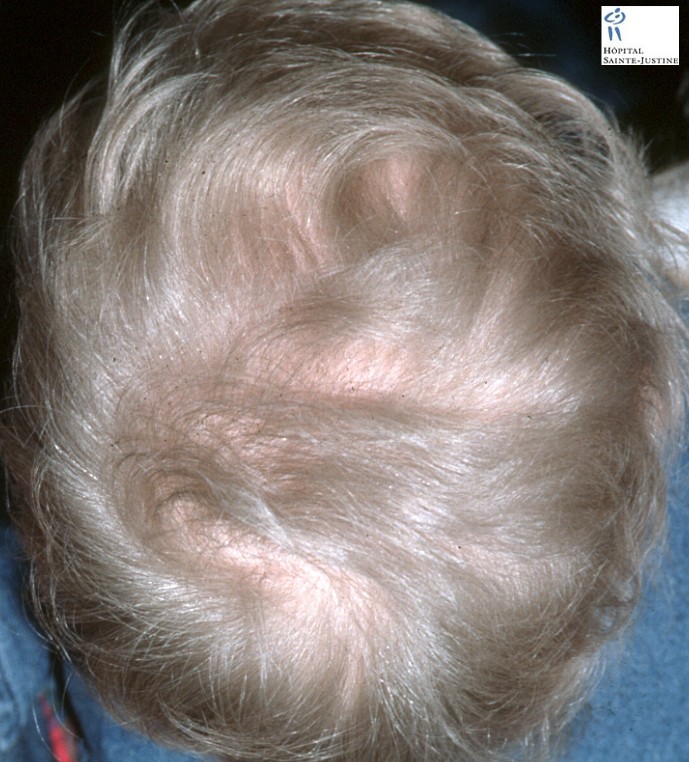
The Griscelli syndrome is characterized by reduced skin pigmentation, often regarded as partial albinism, and silvery-gray hair combined in one type with immunodeficiency.
Three types have been described:
type 1 (MIM.214450) caused by mutations in the myosin VA gene (MYO5A) at chromosome 15q21, and without immunological impairment;
type 2 (MIM.607624) caused by mutations in the RAB27A gene at 15q21, the same location as the MYO5A gene, and with immunological impairment;
type 3 (MIM.609227) characterized by hypomelanosis with no immunological or neurological changes and caused by mutations in the melanophilin (MLPH) gene at 2q37, or the MYO5A gene.
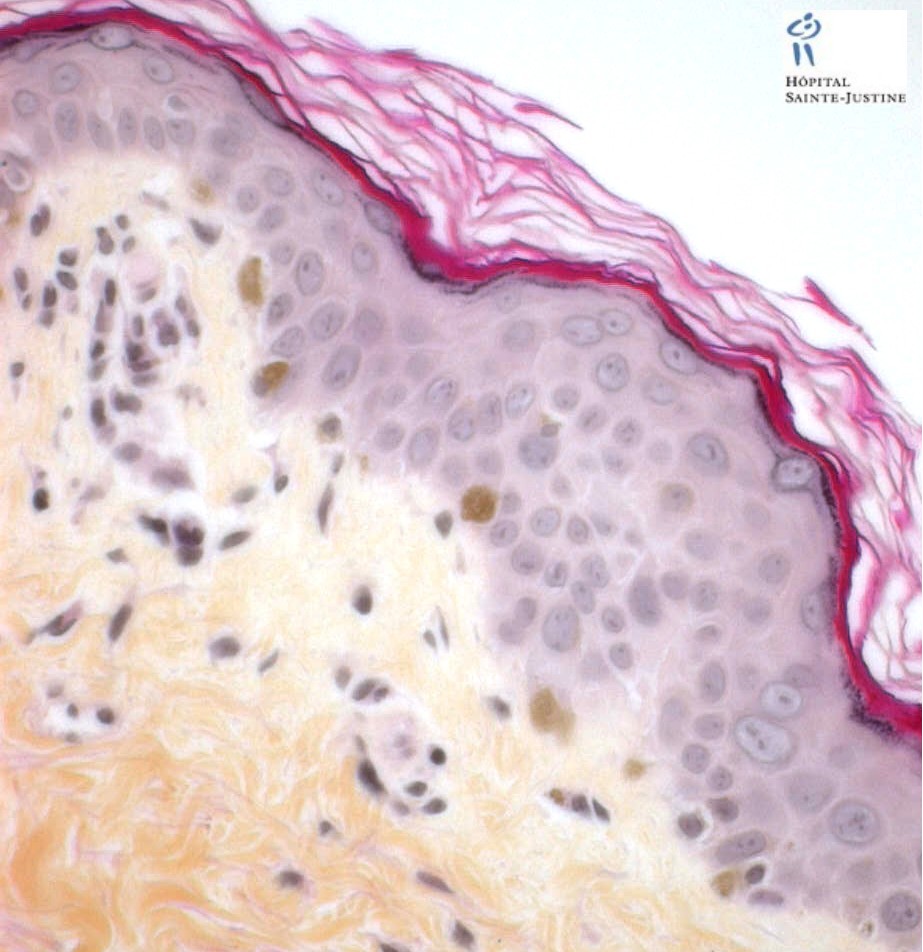
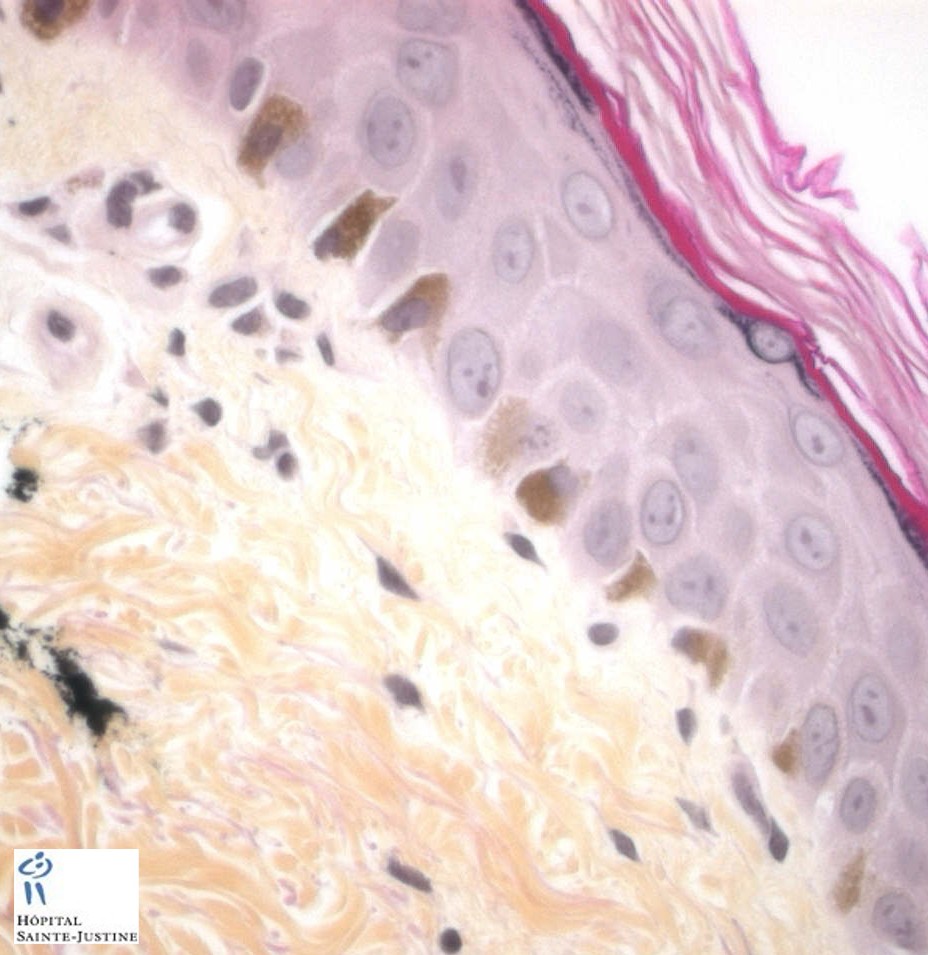
Hisopathology
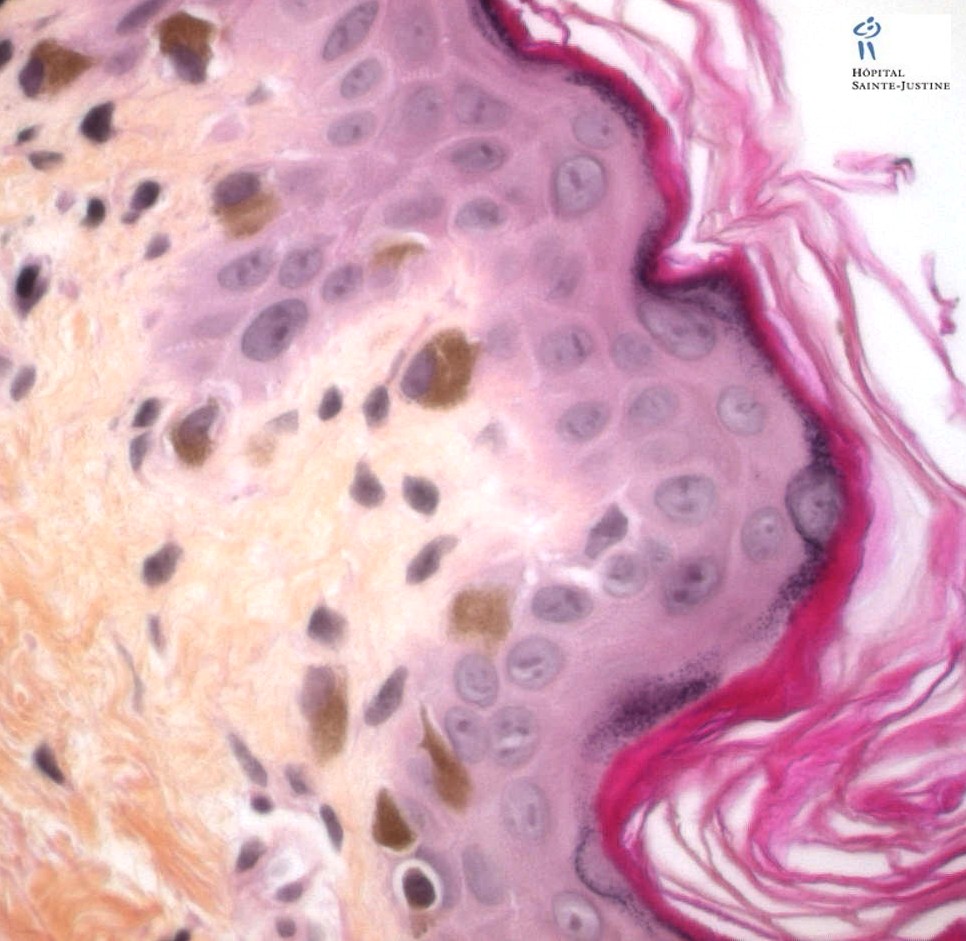
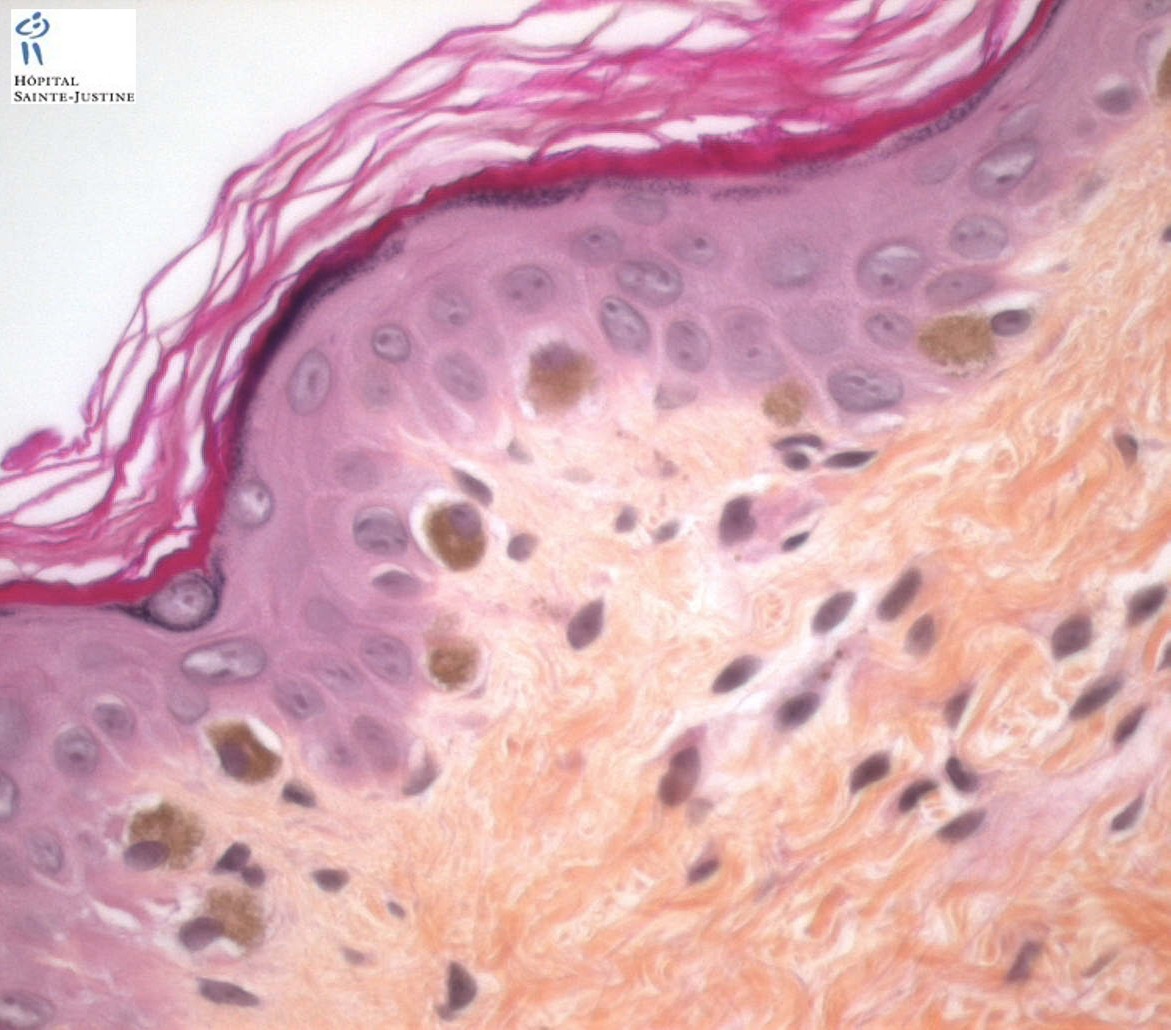
Enlarged hyperpigmented basal melanocytes with sparsely pigmented adjacent keratinocytes are seen on skin biopsy specimens.
There are no abnormal cytoplasmic granules in leukocytes as found in the Chediak–Higashi syndrome.
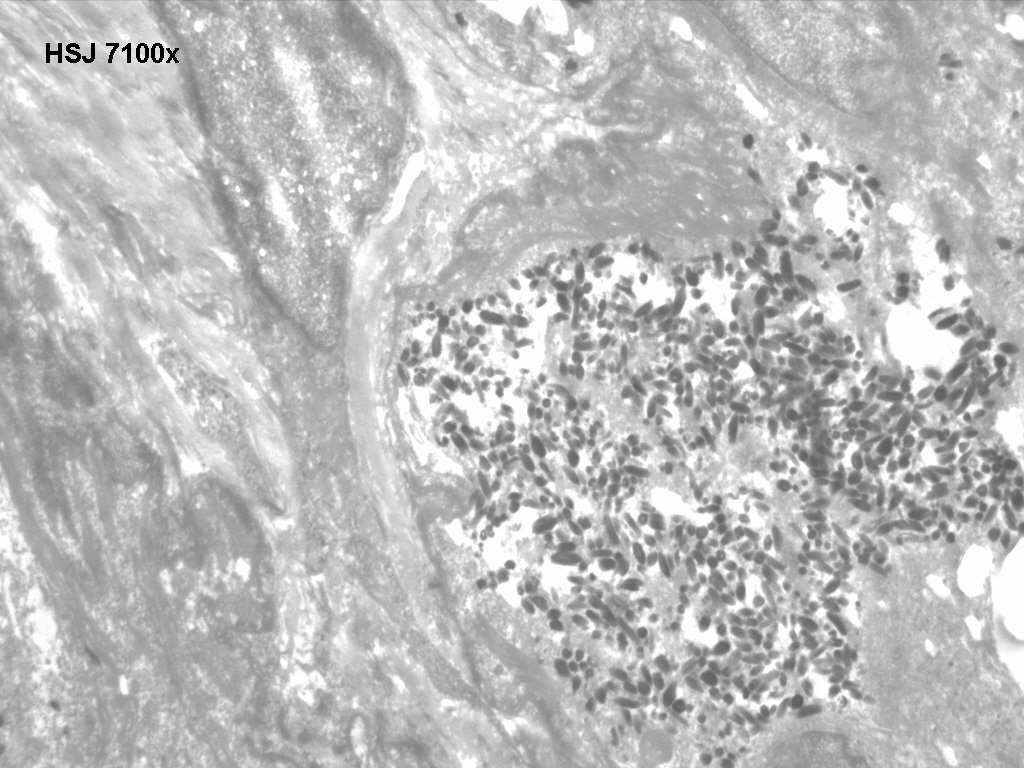
Ultrastructure
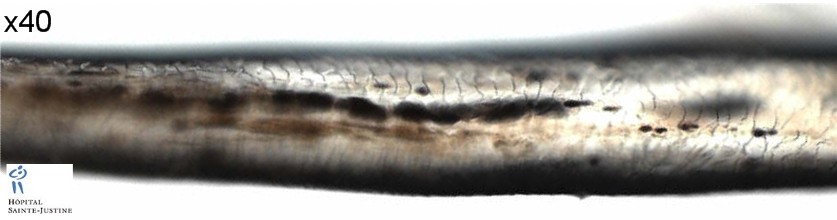
There are some type IV melanosomes in basal melanocytes and shortened dendritic processes. The hair shafts show uneven clusters of aggregated melanin pigment, mainly in the medulla.
Etiology
Mutations in in RAB27A (15q), which codes for a small GTPase(hemophagocytic syndrome)
Mutations in MYO5A, which codes for an actin-based molecular motor (neurological involvement)
The RAB27A and MYO5A gene products interact with each other and function in vesicle trafficking.
Physiopathology
Griscelli disease is a defect in cytoskeletal proteins involved in membrane transport. Griscelli disease, whose clinical manifestations are similar to those of the Chédiak-Higashi disease, is caused by mutations in genes MYO5A (myosin-Va) and RAB27A, a RABs protein.
Unconventional myosins of several classes have been implicated as cytoskeletal proteins involved in actin-dependent movement of organelles and membrane transport in a variety of organisms.
Studies of mice lacking myosin-Va have indicated that it is involved in distributing melanosomes to the cell periphery and in the local movement or processing of organelles in specific regions of neurites.
This finding sheds light on the cause of the pigmentary dilution and the neurologic symptoms associated with the disease in humans.
The similarity of the phenotypes of Griscelli disease and the Chédiak-Higashi disease indicates that the products of the genes encoding myosin-Va and lysosomal-trafficking regulator may interact physically or function in the same intracellular transport pathways responsible for the normal dynamics of lysosome-related organelles.
References
Hong W. Cytotoxic T lymphocyte exocytosis: bring on the SNAREs! Trends Cell Biol. 2005 Dec;15(12):644-50. PMID: 16260137
Stinchcombe J, Bossi G, Griffiths GM. Linking albinism and immunity: the secrets of secretory lysosomes. Science. 2004 Jul 2;305(5680):55-9. PMID: 15232098
Griffiths GM. Albinism and immunity: what’s the link? Curr Mol Med. 2002 Aug;2(5):479-83. PMID: 12125813