serpinopathies
Image Gallery
[ (||image_reduire{0,60}|inserer_attribut{alt,Conformational changes of mutated PI}) ]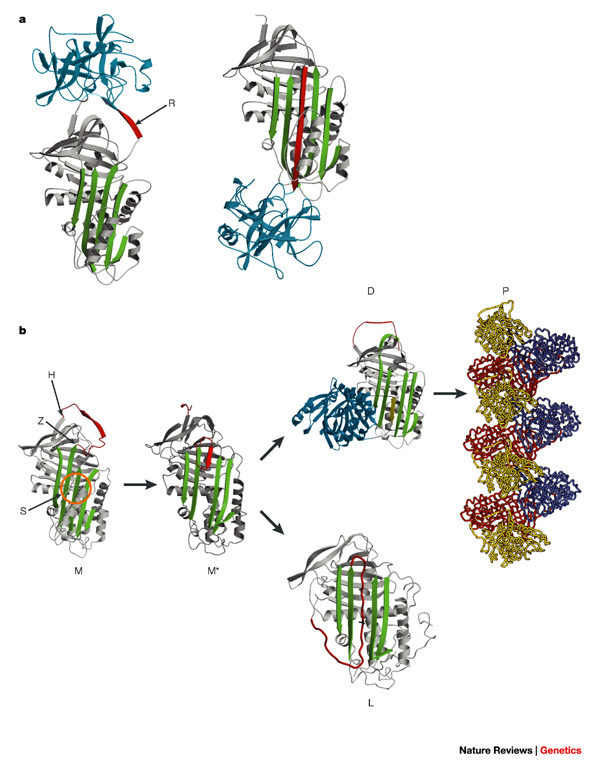{kind=link}
In humans, several serpin mutations are linked to diseases.
Serpins are vulnerable to mutations that result in protein misfolding and the formation of inactive long chain polymers (serpinopathies).
Serpin polymerisation reduces the amount of active inhibitor, as well as accumulation of serpin polymers causing cell death and organ failure.
For example, the serpin alpha1-antitrypsin is primarily produced in the liver, and antitrypsin polymerisation causes hepatocytic necrosis and liver cirrhosis.
The complexity of the serpin mechanism renders these molecules vulnerable to inactivating mutations that promote inappropriate conformational change (or misfolding) and diseases ("serpinopathies").
Well characterised serpinopathies include panlobular emphysema, A1AT-associated cirrhosis, AT3-associated thrombosis and dementia.
Serpin misfolding
Serpins thus belong to a large group of molecules such as the prion proteins and the glutamine repeat containing proteins that are susceptible to misfolding, causing conformational diseases.
The ability to map the mutations in serpins that cause serpinopathies onto a structural framework aided understanding of the mechanism of normal serpin conformational changes, as well as serpin dysfunction.
In particular, many serpin mutations that cause disease localise to two distinct regions of the molecule termed the shutter and the breach. The shutter and the breach contain highly-conserved residues and underlie the path of RCL insertion.
Serpin misfolding results in two common outcomes, both of which stem from the instability of the native (S) conformation. Firstly, pathogenic mutations in serpins can promote inappropriate transition to the monmoeric latent state. This causes disease because it reduces the amount of active inhibitory serpin. For example, the disease-linked antithrombin variants wibble and wobble, both promote formation of the latent state.
Serpin polymerisation
Mutations in serpins may cause polymerisation. While the X-ray crystal structure of an intact serpin polymer remains to be determined, much biochemical, biophysical and structural data suggest that serpins "domain swap" with one another and form long-chain polymers.
This may occur by a RCL of one serpin inserting into the A-sheet of another serpin, to form a chain, rather than inserting into its "own" A-sheet. The polymeric form is inactive and causes pathology.
Serpin polymerisation causes disease in two ways. Firstly, the lack of active serpin results in uncontrolled protease activity and tissue destruction, this is seen in the case of antitrypsin deficiency. Secondly, the polymers themselves clog up the endoplasmic reticulum of cells that synthesize serpins, eventually resulting in cell death and tissue damage.
In the case of antitrypsin deficiency, antitrypsin polymers cause the death of liver cells, eventually resulting in liver damage and cirrhosis.
Finally, it is worth highlighting a structure of a disease-linked human antichymotrypsin variant that demonstrates the extraordinary flexibility of the serpin scaffold.
The structure of antichymotrypsin (Leucine 55 to Proline) revealed a novel "delta" conformation that may represent an intermediate between the native and latent state.
In the delta conformation four residues of the RCL are inserted into the top of β-sheet A. The bottom half of the sheet is filled as a result of one of the α-helices (the F-helix) partially switching to a strand-like conformation, completing the β-sheet hydrogen bonding.
It is unclear whether other serpins can adopt this conformer, or whether this conformation has a functional role. However, this conformation may be important for thyroxine release by Thyroxine binding globulin.
In humans, simple deficiency of many serpins (e.g. through a null mutation) may result in disease.
Rarely, single amino acid changes in the RCL of a serpin alters the specificity of the inhibitor and allow it to target the wrong protease. For example, the Antitrypsin-Pittsburgh mutation (methionine 358 to arginine) allowed the serpin to inhibit thrombin, thus causing a bleeding disorder.
Serpins are suicide inhibitors, the RCL acting as a "bait". Certain disease-linked mutations in the RCL of human serpins permit true substrate-like behaviour and cleavage without complex formation.
Such variants are speculated to affect the rate or the extent of RCL insertion into the A-sheet. These mutations effectively result in serpin deficiency through a failure to properly control the target protease.
Several non-inhibitory serpins play key roles in important human diseases. Most notably, maspin functions as a tumour suppressor in breast and prostate cancer. The mechanism of maspin function remains to be fully understood. Murine knockouts of maspin are lethal; these data suggest that maspin plays a key role in development.
A disadvantageous gain of function
A defining characteristic of conformational diseases is that they arise from mutations that result in a disadvantageous gain of function. Such mutations have consequences over and above any accompanying loss of function.
So, the liver cirrhosis that is associated with alpha1-antitrypsin deficiency arises from conformationally destabilizing mutations of alpha1-antitrypsin, but not from the null mutations that result in total non-expression.
Similarly, whereas non-expressing null variants of antithrombin result in a predisposition to thrombosis, an even greater threat of thrombosis occurs with conformationally destabilizing variants.
These carry with them, as well as plasma deficiency, the risk of pathological monomeric and polymeric conformational transitions that inactivate the protein, with an accompanying sudden and severe onset of thrombosis.
Site specificity of conformational mutations
Another characteristic of conformationally destabilizing mutations is their site specificity. This is seen in the serpins: some 50 such mutations are specifically located in the key mobile regions of these molecules - the hinges of the reactive loop, and the shutter region that underlies the focal opening of the beta-sheet A.
The prime example of a hinge mutation is seen with the replacement, in Z alpha1-antitrypsin, of the conserved glutamate of the proximal hinge of the reactive loop by lysine. The special significance of this replacement, in conformational terms, has been borne out by the subsequent identification of the identical mutation in different situations.
The first was in a family with a deficiency of the plasma anticoagulant heparin cofactor II ; the second was the mutation in Drosophila that is responsible for the serpin dysfunction that produces constitutive activation of antifungal peptides and epidermal necrosis - the necrotic (Nec) phenotype.
However, the most frequent site of destabilizing mutations in the serpins is in the shutter region, which is formed by the closely packed amino-acid side-chains on which the sliding movement of the strands of the beta-sheet A takes place.
Mutations in the strongly conserved region at the commencement of the B-helix in alpha1-antitrypsin, neuroserpin, antithrombin, C1 inhibitor and alpha1-antichymotrypsin all result in polymerization and disease.
The significance of the shutter region, and in particular of its highly conserved core sequence (amino-acid residues 51-56 on the serpin template numbering), was shown by the finding that two other mutants of alpha1-antitrypsin were associated with plasma deficiency and hepatic inclusions: alpha1-antitrypsin Siiyama (S53F) and alpha1-antitrypsin Mmalton (52F deleted).
The Siiyama mutation is the most common cause of severe alpha1-antitrypsin deficiency in Japan and the Mmalton (also known as Mnichinan and Mcagliari) variant is the commonest cause of severe alpha1-antitrypsin deficiency in Sardinia.
Both mutations cause polymerization and the retention of 1-antitrypsin in hepatocytes. Similar mutations in the shutter region of antithrombin (P54T, P54S, S56N) favour the spontaneous formation of polymers and the retention of antithrombin in hepatocytes, with a consequently increased risk of thrombosis.
Other shutter mutations in C1 inhibitor (F52S, P54L) and alpha1-antichymotrypsin (L55P) also cause polymerization and the retention of protein in the liver.
The reduction in the levels of circulating protein in individuals with deficiencies in C1q inhibitor and alpha1-antichymotrypsin allows the uncontrolled activity of proteinases in the complement and inflammatory cascades and hence the clinical syndromes of angio-oedema and panlobar emphysema.
The other vulnerable site for mutations in the serpins is the distal hinge of the reactive-centre loop. Numerous mutations here, especially in antithrombin and C1 inhibitor, result in a thermally sensitive instability that is seen clinically as episodes of thrombosis or angio-oedema triggered by incidental infection and fever.
As well as these main regions of vulnerability, mutations in less crucial regions of the molecule can also predispose to polymer formation, as with the S (E264V) and I (R39C) variants of alpha1-antitrypsin. The point mutations that are responsible for these variants have less effect on beta-sheet A than does the Z variant.
So, the rates of polymer formation are much slower than for Z alpha1-antitrypsin, and results in less retention of protein in hepatocytes, milder plasma deficiency and the lack of a clinical phenotype.
However, if a mild, slowly polymerizing S or I variant of alpha1-antitrypsin is inherited with a rapidly polymerizing Z variant, then the two can interact to form heteropolymeric inclusions in hepatocytes and associated cirrhosis.
Neuroserpin and dementia
The relevance of studies of alpha1-antitrypsin to the dementias was shown in a striking way by the identification of a novel inclusion body dementia called familial encephalopathy with neuroserpin inclusion bodies, or FENI.
This is an autosomal-dominant dementia that is characterized by eosinophilic neuronal inclusions of neuroserpin (Collins’ bodies) in the deeper layers of the cerebral cortex and the substantia nigra.
The inclusions are PAS positive and diastase resistant, and they bear a striking resemblance to those of Z alpha1-antitrypsin that form in the liver.
The observation that FENIB was associated with a mutation, S49P (S53P on the 1-antitrypsin template), in the neuroserpin gene that was homologous to one in alpha1-antitrypsin that causes cirrhosis (S53F) strongly indicated a common molecular mechanism.
This was confirmed by the finding that the neuronal inclusion bodies of FENIB were formed by entangled neuroserpin polymers that were identical in morphology to those from inclusion bodies present in hepatocytes from a child with alpha1-antitrypsin-deficiency-related cirrhosis. Moreover, recombinant S49P neuroserpin polymerizes much faster than the wild-type protein.
The direct relationship between the magnitude of the intracellular accumulation of neuroserpin and the severity of disease has been clearly shown by the recent identification of other neuroserpin mutations in families with FENIB.
In the original family, with the S49P neuroserpin mutation, the affected family members had diffuse, small, intraneuronal inclusions of neuroserpin, and an onset of dementia between the ages of 45 and 60 years.
In a second family, with a conformationally more severe mutation (S52R) and larger inclusions, the onset of dementia was in early adulthood. In a third family, with yet another mutation (H338R), there were more inclusions than for the S52R mutation, and the onset of dementia took place in adolescence. However, the most striking example was the family with the most ’polymerogenic’ mutation of neuroserpin, G392E.
This replacement of a consistently conserved residue in the shutter region resulted in large, multiple inclusions in every neuron, with affected family members dying by the age of 20 years.
So, FENIB shows a clear genotype-phenotype correlation, with the severity of disease correlating closely with the propensity of the mutated neuroserpin to form polymers.
These findings, in addition to previous evidence of the correlation between the polymerization rate of alpha1-antitrypsin mutants and cirrhosis, strongly indicate that intracellular protein aggregation is by itself sufficient to cause neurodegeneration. The role of post-translational degradation of neuroserpin polymers in the development of neurotoxicity has yet to be determined.
Understanding serpinopathies also provides insights on protein misfolding diseases (conformational diseases) as late-onset dementia, amyloidoses, prion encephalopathies and Creutzfeld-Jakob disease (CJD), Huntington disease, Alzheimer disease (AD).
Viral serpins
The viral serpin crmA is a suppressor of the inflammatory response through inhibition of IL-1 and IL-18 processing by the cysteine protease caspase-1 (CASP1).
Polymers and amyloid
The shared feature of the conformational dementias is the formation of intermolecular linkages with resultant protein aggregation. Although the molecular mechanisms involved are diverse and characteristic of each disease, almost all result in beta-linkages formed by hydrogen bonding between peptide loops and sheets.
Some larger, highly ordered proteins, such as the cystatins, can form intermolecular linkages by domain swapping, as with the loop-sheet linkages of the serpins.
The result is the formation of polymers in which the individual molecules substantially retain their ordered structure. However, in other proteins, such as beta2-microglobulin, lysozyme (LYZ) and transthyretin (TTR), linkage occurs by the remarkable realignment of peptide segments to give the sequential layering of -structures known as amyloid.
The characteristic features of amyloid are its staining with the dye Congo red and its birefringence to polarized light.
The determinant feature of amyloid is the formation of a specific pattern on X-ray diffraction, which has been interpreted as being due to the formation of layered arrays of extended beta-sheets.
Amyloid is most frequently observed as large tissue or pericellular deposits; in several systemic amyloidoses this end product interferes with organ function (for example, in the heart, lung or liver) and so is directly responsible for the disease pathology.
The dementias, however, arise specifically from the cumulative loss of neurons, and the pathology is likely to occur directly at the cellular level. Attention is therefore focused on the earliest forms of intracellular association, at the level of fibrils and oligomeric fibrillization intermediates (protofibrils), as well as the preceding dimers and oligomers.
The oligomers also have a predominantly beta-sheet structure and this change in structure underlies neurotoxicity. This is underscored by studies of mutants of alpha-synuclein — a highly conserved protein of 140 amino acids that is expressed predominantly in neurons and is particularly abundant in presynaptic terminals.
It has been suggested that it might have a role in synaptic plasticity and might modulate dopaminergic neuro-transmission. Two amino-acid substitutions in alpha-synuclein, A53T and A30P, are associated with autosomal-dominant, early-onset Parkinson disease.
The mutant protein accumulates as intracytoplasmic inclusions in neurons to form the Lewy bodies that are characteristic of this condition.
Structural studies have shown that the A30P mutant favours the formation of oligomers rather than fibrils, and that these small intermediates could be the toxic species that cause cell death and the associated neurodegenerative disease.
Propagation and prions
Structural studies of the serpins have also shown the feasibility of another perplexing feature of some of the familial and acquired encephalopathies: the ability of the underlying protein oligomers and filaments to self-propagate and even, with the prions, to propagate infectively.
Aggregation to form polymeric filaments is particularly likely to occur when there is a beta-strand receptor, as in the main beta-sheet of the conformationally unstable variants of alpha1-antitrypsin.
Both the serpins and the prions readily dimerize by domain swapping, and the initial oligomers, like those formed by serpins, act as a template for propagation of the conformational change.
The change is propagated from molecule to molecule, which then extend to give long-chain polymers. This is well illustrated by mutations of antithrombin.
Whereas mutations in alpha1-antitrypsin and neuroserpin result in the formation of long-chain polymers, those in antithrombin result in the formation of an inactive antithrombin monomer.
Moreover, in a process similar to that proposed for the prion encephalopathies, this aberrant form of antithrombin then binds to a normal antithrombin molecule, which leads to the propagation of conformational inactivation.
A more direct insight into the mechanisms of prion propagation that underlie the spongiform encephalopathies is provided by studies of the unrelated yeast prion Ure2. This normally soluble and highly ordered molecule undergoes a conformational change to form fibrils that bind the dye Congo red and show typical amyloid-like birefringence.
However, these fibrils do not have the cross-beta-structure of amyloid and, as is also observed with fibrils formed by orderly aggregation of serpins, the component molecules of the fibril retain their helical structure.
These amyloid-like fibrils are self-propagating but lose this ability over time, coincident with a transition of the fibril to give the typical amyloid X-ray fibre diffraction pattern.
These findings fit with a range of others and together they indicate that the neuronal pathology of the conformational dementias results from the early stages of intracellular aggregation, before or incidental to amyloid formation.
See also
![]() proteases inhibitors (PIs)
proteases inhibitors (PIs)
![]() serpins
serpins
![]() conformational diseases
conformational diseases
References
![]() Lomas DA, Carrell RW. Serpinopathies and the conformational dementias. Nat Rev Genet. 2002 Oct;3(10):759-68. PMID: #12360234#
Lomas DA, Carrell RW. Serpinopathies and the conformational dementias. Nat Rev Genet. 2002 Oct;3(10):759-68. PMID: #12360234#