celiac disease
Image Gallery
[ (||image_reduire{0,60}|inserer_attribut{alt,33-mer and celiac disease}) ]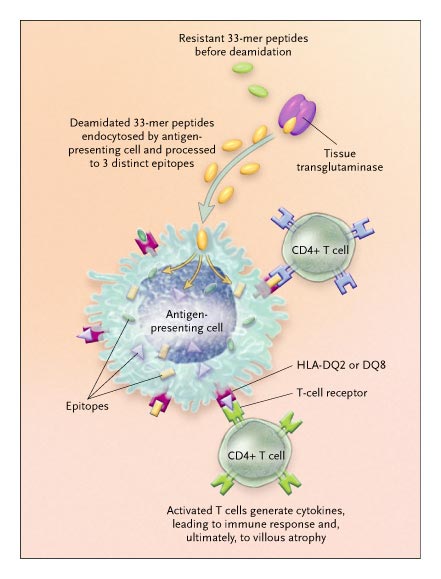{kind=link}
Celiac disease (also known as celiac sprue and gluten-sensitive enteropathy) is a common autoimmune condition triggered by ingesting one of several related proteins found in wheat, barley, and rye: the gliadins, hordeins, and secalins.
In susceptible persons, ingestion of these proteins leads to infiltration of the intestinal mucosa by both intraepithelial CD8+ lymphocytes and CD4+ lamina propria lymphocytes and, ultimately, to crypt hyperplasia and villous atrophy.
Symptoms vary — malabsorption of food by the intestine, diarrhea, and failure to thrive are typical in affected children, and symptoms in adults can include depression and anemia.
A gluten-free diet alleviates these symptoms, although adherence to such a diet can be difficult. Shan and colleagues3 have recently identified a peptide that probably initiates the disease, raising the possibility that strategic inroads can be made into the disorder.
Synopsis
![]() intestinal villous atrophy
intestinal villous atrophy
![]() increase of intraepithelial lymphocytes
increase of intraepithelial lymphocytes
![]() intestinal crypt hyperplasia
intestinal crypt hyperplasia
![]() chorionic lymphoplasmocytic infiltration
chorionic lymphoplasmocytic infiltration
Differential diagnosis
![]() other causes of intestinal villous atrophy
other causes of intestinal villous atrophy
![]() enteropathy-type T-cell lymphoma
enteropathy-type T-cell lymphoma
Associations
![]() autoimmune diabetes
autoimmune diabetes
![]() autoimmune hepatitis
autoimmune hepatitis
![]() systemic autoimmune disorders (#16133974#)
systemic autoimmune disorders (#16133974#)
Marsh classification of intestinal villous atrophy
| Types | Type 0 | Type 1 | Type 2 | Type 3a | Type 3b | Type 3c |
| IEL | <40 | >40 | >40 | >40 | >40 | >40 |
| Crypts | Normal | Normal | Hypertrophic | Hypertrophic | Hypertrophic | Hypertrophic |
| Villi | Normal | Normal | Normal | Mild atrophy | Marked atrophy | Absent |
NB: IEL, IntraEpithelial Lymphocytes (per 100 epithelial cells)
Physiopathology
There is increasing evidence that CD4+ T cells mediate the pathogenic process in celiac disease.
First, the principal determinants of genetic susceptibility are the highly variable HLA class II DQA and DQB genes located in the major histocompatibility complex. These genes (specifically the combination of variant alleles HLA-DQA1*0501 and DQB1*0201) encode the HLA-DQ2 class II protein molecule, which presents peptides to and binds CD4. A less prevalent determinant of susceptibility is the HLA-DQ8 variant.
Second, HLA-DQ2–restricted T-cell clones that are specific for gliadin have been isolated from the small intestines of patients with celiac disease. However, these clones produce only small amounts of cytokines, and the mechanism by which gliadin peptides bind with high affinity to the HLA-DQ2–binding groove has only recently become clear.
The presence of endomysial autoantibody is another indicator of celiac disease, and the identification of tissue transglutaminase as the target of this antibody4 has been enlightening. This enzyme is expressed on the subepithelial layer of intestinal epithelium, where it deamidates the glutamine residues in gliadin, resulting in glutamic acids. Deamidated peptides adhere strongly to the binding grooves of HLA-DQ2 and DQ8 molecules and elicit strong T-cell responses.5
Although the ingested proteins responsible for celiac disease may carry epitopes capable of activating T cells, they are also substrates for proteolytic degradation by gastrointestinal enzymes and thus should be fully digested before any exposure to the immune system could possibly occur.
Therefore, the question of whether peptides constituting T-cell epitopes can survive the degradations of a low pH and proteolytic enzymes has critical implications for their functional relevance.
One 33-amino-acid (33-mer) peptide survives transit through the digestive enzymatic milieu and arrives intact in the small intestine. In a series of elegant experiments, they demonstrated that this 33-mer resists digestion by gastric and intestinal proteolytic enzymes for extended periods in vitro.
The peptide is resistant — both in vitro and in vivo — to digestion by brush-border enzymes of the small intestinal mucosa of rats and humans, although these enzymes normally reduce any remaining peptides to single amino acids or small peptides of about two or three residues before they are absorbed.
The 33-mer carries multiple copies of three epitopes that are immunogenic in patients with celiac disease. Furthermore, the 33-mer has a very high affinity for tissue transglutaminase. Shan et al. found that once it was deamidated by tissue transglutaminase, the 33-mer elicited a response from each of 14 polyclonal T-cell lines derived from different patients with celiac disease.
It therefore has many, if not all, of the properties required to initiate a response in patients with celiac disease. It survives the digestive tract, is a good substrate for tissue transglutaminase, is loaded onto HLA-DQ molecules, and activates T cells — which may then drive the characteristic immune response in the small intestinal mucosa. Similar peptide sequences are present in the hordeins and secalins.
The 33-amino-acid (33-mer) peptide arrives intact in the small intestine, where it undergoes deamidation by tissue transglutaminase.
By means of endocytosis, the deamidated peptide then enters the antigen-presenting cell, where it is processed to three epitopes that bind to the HLA-DQ2 or DQ8 molecule and are subsequently recognized by T-cell receptors of CD4+ T cells.
The activated CD4+ T cells generate cytokines, prompting the immune response and, ultimately, the villous atrophy and crypt hyperplasia that are characteristic of celiac disease.
Of potential therapeutic consequence is the finding that the 33-mer is broken down by a bacterial prolyl endopeptidase, raising the encouraging possibility of alternatives to a gluten-free diet (perhaps including genetic modification of the offending sequence) for the treatment of celiac disease. Successful clinical trials of such peptidases would provide final proof of the hypothesis that the 33-mer is central to the molecular pathologic process of celiac disease.
Despite these welcome findings, many questions remain. For example, up to 30 percent of persons of North European ancestry express HLA-DQ2, but celiac disease develops in only a small proportion of these carriers.
Although there is a pronounced familial aggregation of celiac disease, the pattern of inheritance is complex. Hence, it is clear that other genetic and possibly environmental influences have yet to be identified.
The role of intraepithelial CD8+ lymphocytes (which do not bind class II molecules of the major histocompatibility complex) in the pathogenesis of the disease also remains to be determined.
These questions notwithstanding, the identification of a pathogenetic pathway involving autoantibodies and cell-based immunity in celiac disease demonstrates the importance of research on principles of immunology and offers new hope for the understanding of other complex disorders.
Genetic predisposition
Coeliac disease is an inflammatory disorder of the small intestine with an autoimmune component and strong heritability. Genetic studies have confirmed strong association to HLA and identified 39 non-HLA risk genes, mostly immune-related.
Over 50% of the disease-associated single nucleotide polymorphisms are correlated with gene expression.
Most of the coeliac disease-associated regions are shared with other immune-related diseases, as well as with metabolic, haematological or neurological traits, or cancer.
![]() trisomy 21 (#10997372#)
trisomy 21 (#10997372#)
![]() familial celiac disease
familial celiac disease
![]() HLA class II genes explain 40% of the heritable risk.
HLA class II genes explain 40% of the heritable risk.
![]() non-HLA genes accounting for most of the familial clustering
non-HLA genes accounting for most of the familial clustering
- MYO9B gene variant (#16282976#)
Genetic predisposition loci
| CELIAC1 | MIM.604305 | 6p21.3 | |
| CELIAC2 | MIM.609754 | 5q31-q33 | |
| CELIAC3 | MIM.609755 | 2q33 | CTLA4 |
| CELIAC4 | MIM.609753 | 19p13.1 | MYO9B |
| CELIAC5 | MIM.607202 | 15q11-q13 | |
| CELIAC6 | MIM.611598 | 4q27 | |
| CELIAC7 | MIM.612005 | 1q31 | RGS1 |
| CELIAC8 | MIM.612006 | 2q11-q12 | |
| CELIAC9 | MIM.612007 | 3p21 | |
| CELIAC10 | MIM.612008 | 3q25-q26 | IL12A |
| CELIAC11 | MIM.612009 | 3q28 | |
| CELIAC12 | MIM.612010 | 6q25 | |
| CELIAC13 | MIM.612011 | 12q24 |
See also
![]() refractory sprue (refractory celiac disease)
refractory sprue (refractory celiac disease)
![]() enteropathy-associated T-cell lymphoma
enteropathy-associated T-cell lymphoma
References
![]() Oberhuber G, Granditsch G, Vogelsang H. The histopathology of coeliac disease: time for a standardized report scheme for pathologists. Eur J Gastroenterol Hepatol. 1999 Oct;11(10):1185-94. Review.PMID: #10524652#
Oberhuber G, Granditsch G, Vogelsang H. The histopathology of coeliac disease: time for a standardized report scheme for pathologists. Eur J Gastroenterol Hepatol. 1999 Oct;11(10):1185-94. Review.PMID: #10524652#
Reviews
![]() A genetic perspective on coeliac disease. Trynka G, Wijmenga C, van Heel DA. Trends Mol Med. 2010 Nov;16(11):537-50. PMID: #20947431#
A genetic perspective on coeliac disease. Trynka G, Wijmenga C, van Heel DA. Trends Mol Med. 2010 Nov;16(11):537-50. PMID: #20947431#
![]() Green PH, Cellier C. Celiac disease. N Engl J Med. 2007 Oct 25;357(17):1731-43. PMID: #17960014#
Green PH, Cellier C. Celiac disease. N Engl J Med. 2007 Oct 25;357(17):1731-43. PMID: #17960014#
![]() Sollid LM, Jabri B. Is celiac disease an autoimmune disorder? Curr Opin Immunol. 2005 Dec;17(6):595-600. PMID: #16214317#
Sollid LM, Jabri B. Is celiac disease an autoimmune disorder? Curr Opin Immunol. 2005 Dec;17(6):595-600. PMID: #16214317#
![]() Treem WR. Emerging concepts in celiac disease. Curr Opin Pediatr. 2004 Oct;16(5):552-9. PMID: #15367850#
Treem WR. Emerging concepts in celiac disease. Curr Opin Pediatr. 2004 Oct;16(5):552-9. PMID: #15367850#
![]() Celiac disease—the villain unmasked? McManus R, Kelleher D. N Engl J Med. 2003 Jun 19;348(25):2573-4. PMID: #12815145#
Celiac disease—the villain unmasked? McManus R, Kelleher D. N Engl J Med. 2003 Jun 19;348(25):2573-4. PMID: #12815145#
![]() Farrell RJ, Kelly CP. Celiac sprue. N Engl J Med. 2002 Jan 17;346(3):180-8. PMID: #11796853#
Farrell RJ, Kelly CP. Celiac sprue. N Engl J Med. 2002 Jan 17;346(3):180-8. PMID: #11796853#
![]() Vogelsang H, Schwarzenhofer M, Oberhuber G. Changes in gastrointestinal permeability in celiac disease. Dig Dis. 1998 Nov-Dec;16(6):333-6. PMID: #10207217#
Vogelsang H, Schwarzenhofer M, Oberhuber G. Changes in gastrointestinal permeability in celiac disease. Dig Dis. 1998 Nov-Dec;16(6):333-6. PMID: #10207217#
![]() McManus R, Kelleher D. Celiac disease—the villain unmasked? N Engl J Med. 2003 Jun 19;348(25):2573-4. PMID: #12815145#
McManus R, Kelleher D. Celiac disease—the villain unmasked? N Engl J Med. 2003 Jun 19;348(25):2573-4. PMID: #12815145#
![]() Farrell RJ, Kelly CP. Celiac sprue. N Engl J Med. 2002 Jan 17;346(3):180-8. PMID: #11796853#
Farrell RJ, Kelly CP. Celiac sprue. N Engl J Med. 2002 Jan 17;346(3):180-8. PMID: #11796853#