rhabdomyosarcomas
Image Gallery
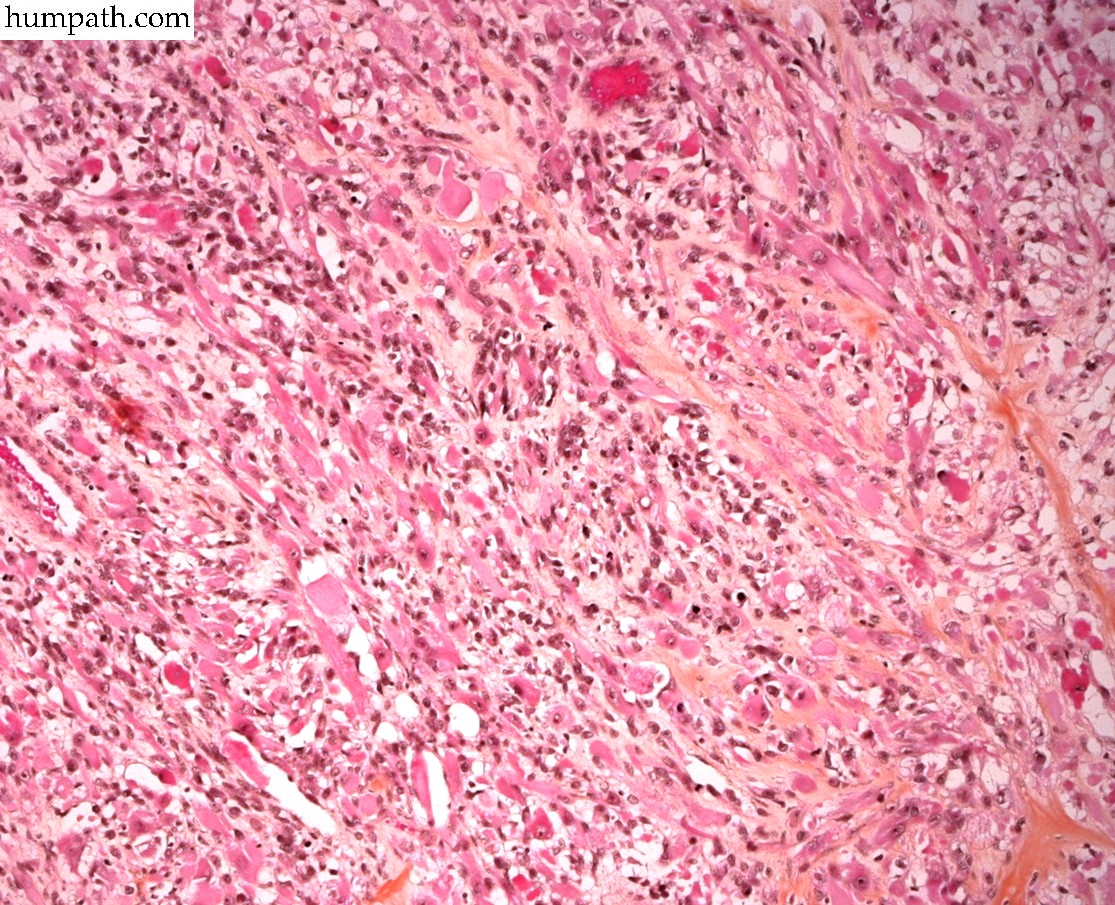
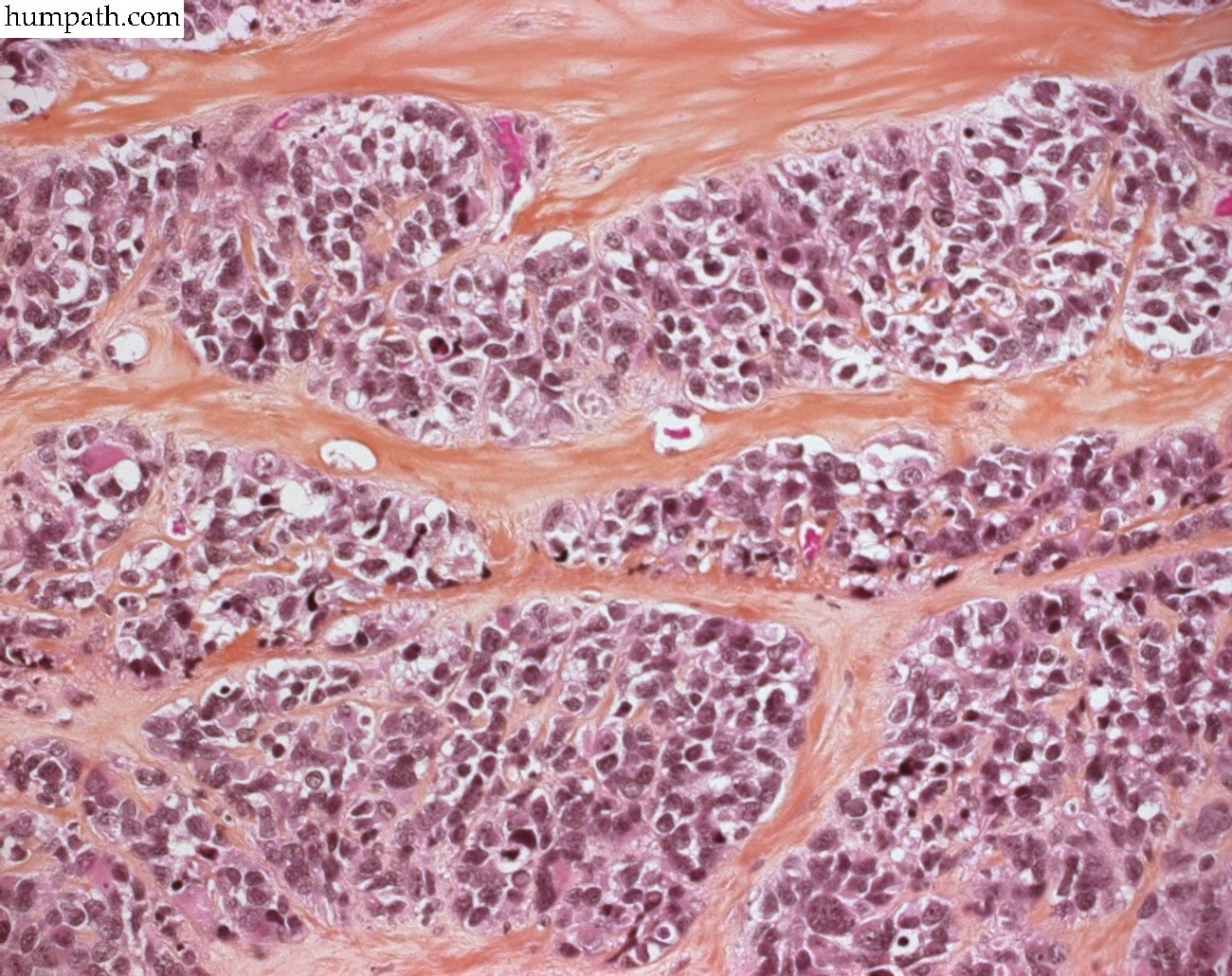
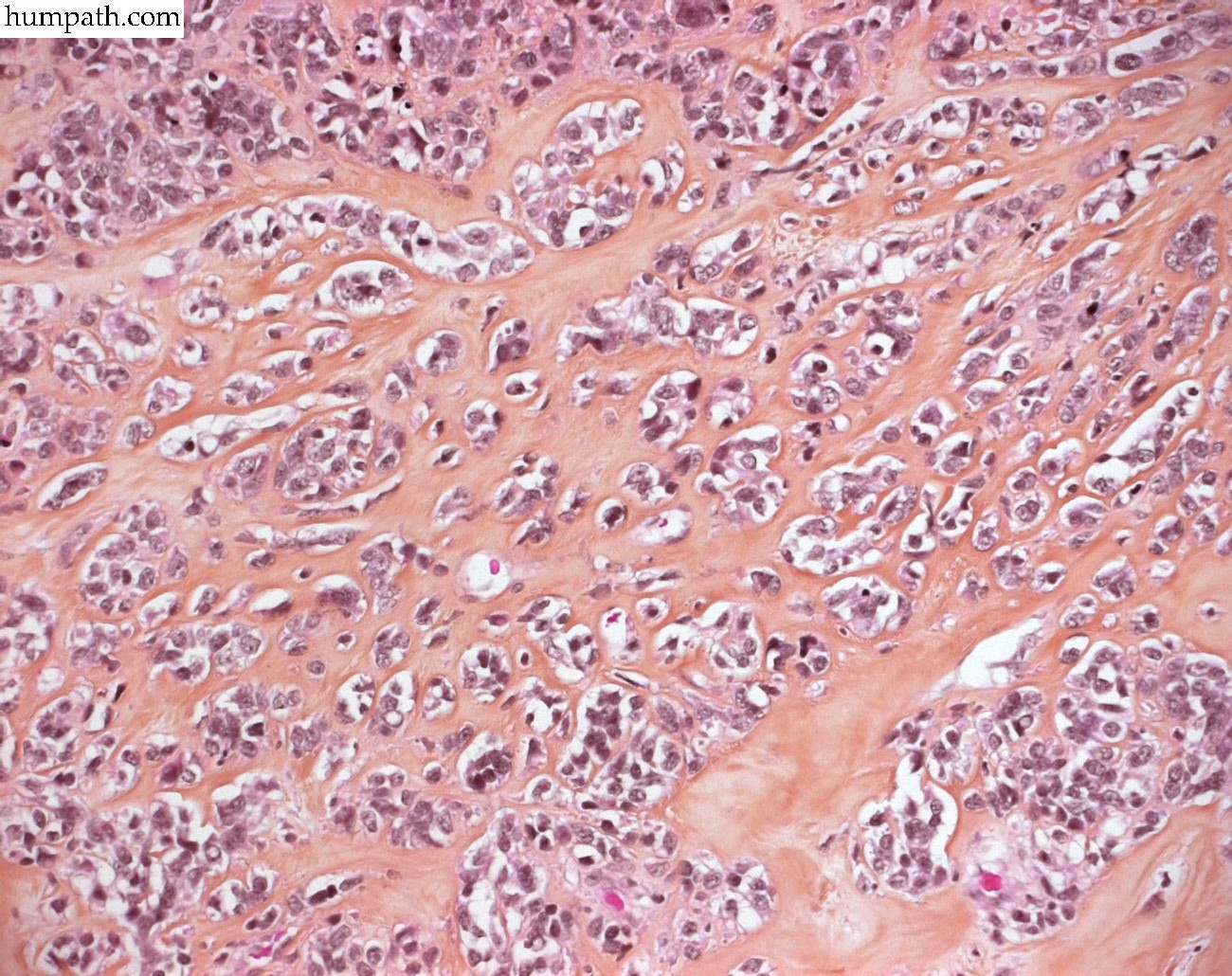
[ (||image_reduire{0,60}|inserer_attribut{alt,Embryonal rhabdomyosarcoma}) ] [ (||image_reduire{0,60}|inserer_attribut{alt,Botryoid embryonal rhabdomyosarcoma of the bladder}) ] [ (||image_reduire{0,60}|inserer_attribut{alt,Botryoid embryonal rhabdomyosarcoma of the bladder}) ] [ (||image_reduire{0,60}|inserer_attribut{alt,Botryoid embryonal rhabdomyosarcoma of the bladder}) ] [ (||image_reduire{0,60}|inserer_attribut{alt,Sclerosing rhabdomyosarcoma}) ] [ (||image_reduire{0,60}|inserer_attribut{alt,Sclerosing rhabdomyosarcoma}) ] [ (||image_reduire{0,60}|inserer_attribut{alt,Sclerosing rhabdomyosarcoma}) ] [ (||image_reduire{0,60}|inserer_attribut{alt,Alveolar rhabdomyosarcoma (leg)}) ] [ (||image_reduire{0,60}|inserer_attribut{alt,Alveolar rhabdomyosarcoma (leg)}) ]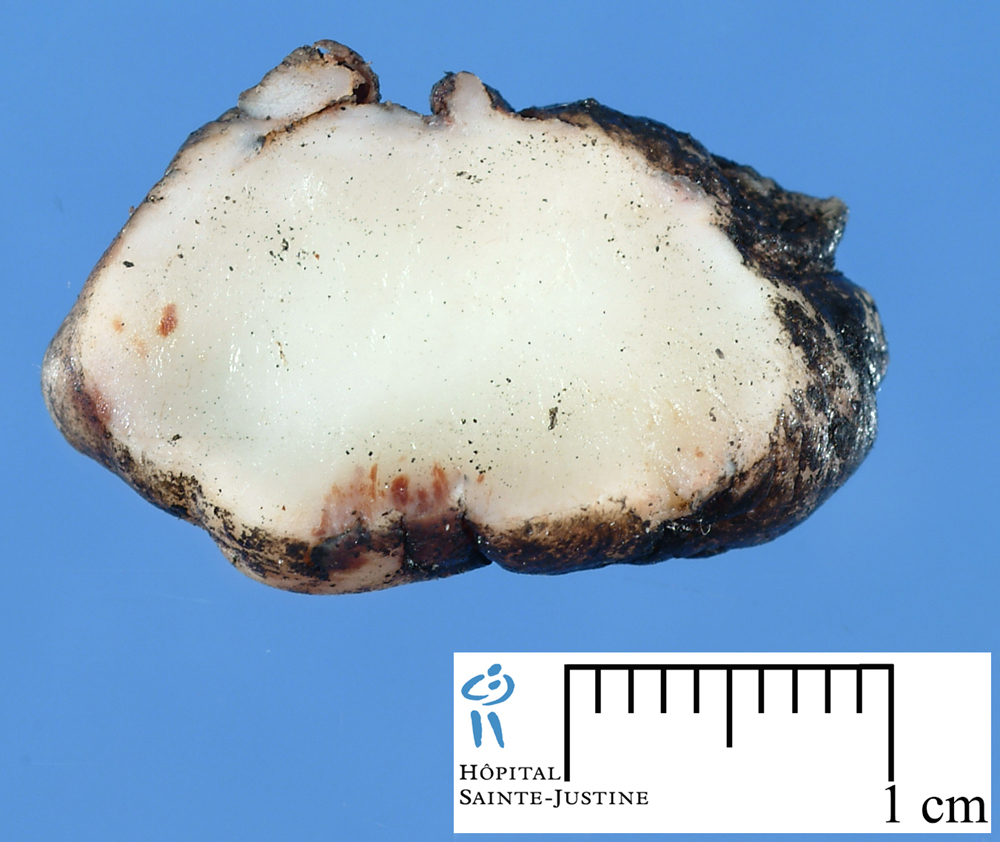{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
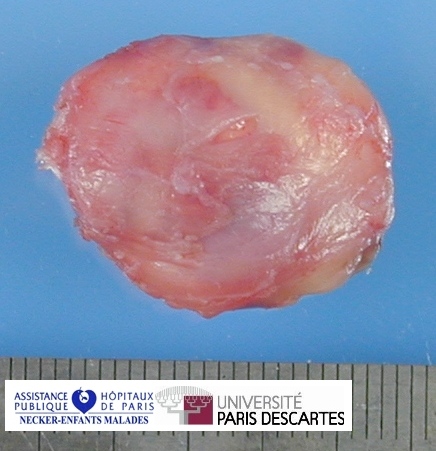{kind=link}
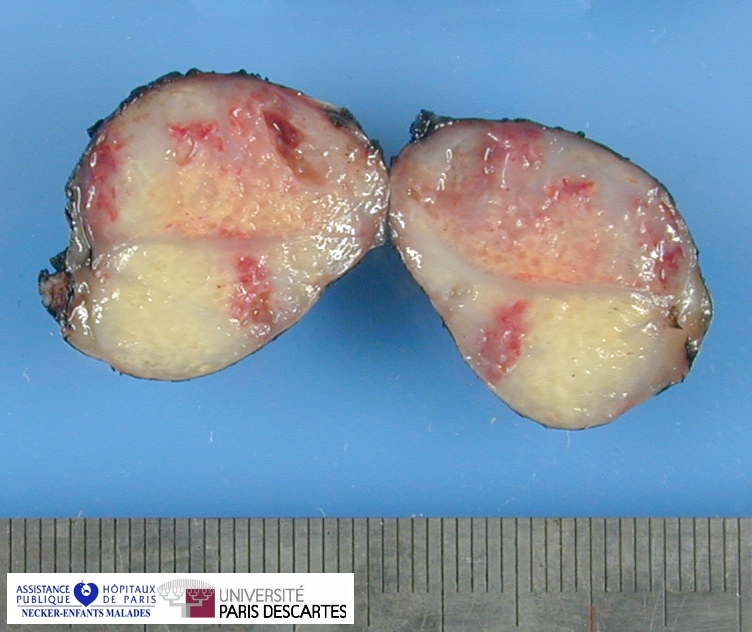{kind=link}
Digital cases
![]() Case 181 (HPC:181) : Embryonal rhabdomyosarcoma of the maxillary
Case 181 (HPC:181) : Embryonal rhabdomyosarcoma of the maxillary
![]() Case 210 (HPC:210) : Embryonal rhabdomyosarcoma
Case 210 (HPC:210) : Embryonal rhabdomyosarcoma
![]() Case 211 (HPC:211) : Botryoid embryonal rhabdomyosarcoma
Case 211 (HPC:211) : Botryoid embryonal rhabdomyosarcoma
![]() Case 215 (HPC:215) : Alveolar rhabdomyosarcoma, solid variant
Case 215 (HPC:215) : Alveolar rhabdomyosarcoma, solid variant
![]() Case 220 (HPC:220) : Alveolar rhabdomyosarcoma
Case 220 (HPC:220) : Alveolar rhabdomyosarcoma
![]() Case 250 : Botryoid embryonal rhabdomyosarcoma of the nasopharynx
Case 250 : Botryoid embryonal rhabdomyosarcoma of the nasopharynx
Definition: Rhabdomyosarcoma (RMS) is the most common soft-tissue sarcoma of childhood. Rhabdomyosarcoma is a soft tissue tumor of striated muscle origin. It is observed mainly in children and adolescents. Rhabdomyosarcoma predominantly occurs in three regions: head and neck, genito-urinary tract and retroperitoneum, and upper and lower extremities.
Rhabdomyosarcoma is the fourth most common solid tumor and the most common soft tissue sarcoma in children. It can occur anywhere, except the brain, as it arises from totipotential mesenchymal cells.
It is an aggressive tumor with local invasion and a tendency for early recurrence if not completely excised.
It is most often seen in children between the ages of two and six years and carries the poorest prognosis when seen in children under twelve months.
The most common histologic type, embryonal rhabdomyosarcoma, offers the best prognosis. Most tumors originate in the pelvis or genitourinary tract (39% of cases) with 31% occurring in the head and neck region.
The treatment is usually chemotherapy with or without radiotherapy and subsequent surgery. The five year survival is 40%.
Differential diagnosis
While, in the past, this distinction was made largely on architectural grounds (i.e. the presence of alveolar spaces), it is nowadays recognized that cytological features are most reliable in this regard—specifically, the cells of alveolar rhabdomyosarcoma have more uniform morphology, are more consistently rounded and have larger nuclei than those of embryonal rhabdomyosarcoma.
In addition, pleomorphic rhabdomyosarcoma, which is virtually confined to adults, is diagnosed somewhat more often nowadays, largely as a result of the implementation of more sensitive and specific immunostains.
Pleomorphic rhabdomyosarcomas are among the most aggressive pleomorphic sarcomas of adulthood.
Types
![]() embryonal rhabdomyosarcoma (ERMS)
embryonal rhabdomyosarcoma (ERMS)
![]() alveolar rhabdomyosarcoma (ARMS)
alveolar rhabdomyosarcoma (ARMS)
![]() sclerosing rhabdomyosarcoma (SRMS)
sclerosing rhabdomyosarcoma (SRMS)
Variants
![]() pleomorphic rhabdomyosarcoma
pleomorphic rhabdomyosarcoma
![]() anaplastic rhabdomyosarcoma (#12550764#, #8333559#, #8470759#)
anaplastic rhabdomyosarcoma (#12550764#, #8333559#, #8470759#)
![]() fusiform cell rhabdomyosarcoma (spindle-cell rhabdomyosarcoma) (#9537474#, #8434703#, #1599014#, #16006807#)
fusiform cell rhabdomyosarcoma (spindle-cell rhabdomyosarcoma) (#9537474#, #8434703#, #1599014#, #16006807#)
Epidemiology
![]() 5-10% of patients with rhabdomyosarcomas (RMS) are diagnosed during the first year of life
5-10% of patients with rhabdomyosarcomas (RMS) are diagnosed during the first year of life
Immunochemistry
![]() ALK +/- (15 to 45%) (#12405914#)
ALK +/- (15 to 45%) (#12405914#)
![]() WT1 + (cytoplasmic) (#12379755#)
WT1 + (cytoplasmic) (#12379755#)
Localization
![]() head and neck
head and neck
- parotid region (#11753993#)
![]() trunk
trunk
![]() biliary tract
biliary tract
![]() pelvis
pelvis
- bladder
![]() paratesticular region
paratesticular region
![]() limbs
limbs
Differential diagnosis
![]() leukemias (#9870849#)
leukemias (#9870849#)
CGH: #11807989#
Gene mutations
![]() rare CDKN2A (p16INK4A/p14ARF) somatic mutations (#17243166#)
rare CDKN2A (p16INK4A/p14ARF) somatic mutations (#17243166#)
Gene overexpression
![]() FGFR1 over-expression in primary rhabdomyosarcoma tumors
FGFR1 over-expression in primary rhabdomyosarcoma tumors
- by FGFR1 amplification (8p11 amplification) in pleomorphic rhabdomyosarcoma (PRMS) (#16790082#)
- by hypomethylation(#17696196#)
Animal model
![]() Patched1 knock-outmouse model has shown that RMS characteristically displays an activation of cell survival promoting and anti-apoptotic proteins such as Igf2 (IGF2), Akt (AKT), and Bcl-2 (BCL2).
Patched1 knock-outmouse model has shown that RMS characteristically displays an activation of cell survival promoting and anti-apoptotic proteins such as Igf2 (IGF2), Akt (AKT), and Bcl-2 (BCL2).
Reviews
![]() Xia SJ, Pressey JG, Barr FG. Molecular pathogenesis of rhabdomyosarcoma. Cancer Biol Ther. 2002 Mar-Apr;1(2):97-104. PMID: #12170781#
Xia SJ, Pressey JG, Barr FG. Molecular pathogenesis of rhabdomyosarcoma. Cancer Biol Ther. 2002 Mar-Apr;1(2):97-104. PMID: #12170781#
References
![]() Goldstein M, Meller I, Orr-Urtreger A. FGFR1 over-expression in primary rhabdomyosarcoma tumors is associated with hypomethylation of a 5’ CpG Island and abnormal expression of the AKT1, NOG, and BMP4 genes. Genes Chromosomes Cancer. 2007 Aug 15; PMID: #17696196#
Goldstein M, Meller I, Orr-Urtreger A. FGFR1 over-expression in primary rhabdomyosarcoma tumors is associated with hypomethylation of a 5’ CpG Island and abnormal expression of the AKT1, NOG, and BMP4 genes. Genes Chromosomes Cancer. 2007 Aug 15; PMID: #17696196#
![]() Chen Y, Takita J, Mizuguchi M, Tanaka K, Ida K, Koh K, Igarashi T, Hanada R, Tanaka Y, Park MJ, Hayashi Y. Mutation and expression analyses of the MET and CDKN2A genes in rhabdomyosarcoma with emphasis on MET overexpression. Genes Chromosomes Cancer. 2007 Apr;46(4):348-58. PMID: #17243166#
Chen Y, Takita J, Mizuguchi M, Tanaka K, Ida K, Koh K, Igarashi T, Hanada R, Tanaka Y, Park MJ, Hayashi Y. Mutation and expression analyses of the MET and CDKN2A genes in rhabdomyosarcoma with emphasis on MET overexpression. Genes Chromosomes Cancer. 2007 Apr;46(4):348-58. PMID: #17243166#
![]() Parham DM, Ellison DA. Rhabdomyosarcomas in adults and children: an update. Arch Pathol Lab Med. 2006 Oct;130(10):1454-65. PMID: #17090187#
Parham DM, Ellison DA. Rhabdomyosarcomas in adults and children: an update. Arch Pathol Lab Med. 2006 Oct;130(10):1454-65. PMID: #17090187#
![]() Wachtel M, Runge T, Leuschner I, Stegmaier S, Koscielniak E, Treuner J, Odermatt B, Behnke S, Niggli FK, Schafer BW. Subtype and prognostic classification of rhabdomyosarcoma by immunohistochemistry. J Clin Oncol. 2006 Feb 10;24(5):816-22. PMID: #16391296#
Wachtel M, Runge T, Leuschner I, Stegmaier S, Koscielniak E, Treuner J, Odermatt B, Behnke S, Niggli FK, Schafer BW. Subtype and prognostic classification of rhabdomyosarcoma by immunohistochemistry. J Clin Oncol. 2006 Feb 10;24(5):816-22. PMID: #16391296#
![]() Qualman SJ, Bowen J, Parham DM, Branton PA, Meyer WH; Members of the Cancer Committee, College of American Pathologists. Protocol for the examination of specimens from patients (children and young adults) with rhabdomyosarcoma. Arch Pathol Lab Med. 2003 Oct;127(10):1290-7. PMID: #14521467#
Qualman SJ, Bowen J, Parham DM, Branton PA, Meyer WH; Members of the Cancer Committee, College of American Pathologists. Protocol for the examination of specimens from patients (children and young adults) with rhabdomyosarcoma. Arch Pathol Lab Med. 2003 Oct;127(10):1290-7. PMID: #14521467#
![]() Bridge JA, Liu J, Qualman SJ, Suijkerbuijk R, Wenger G, Zhang J, Wan X, Baker KS, Sorensen P, Barr FG. Genomic gains and losses are similar in genetic and histologic subsets of rhabdomyosarcoma, whereas amplification predominates in embryonal with anaplasia and alveolar subtypes. Genes Chromosomes Cancer. 2002 Mar;33(3):310-21. PMID: #11807989#
Bridge JA, Liu J, Qualman SJ, Suijkerbuijk R, Wenger G, Zhang J, Wan X, Baker KS, Sorensen P, Barr FG. Genomic gains and losses are similar in genetic and histologic subsets of rhabdomyosarcoma, whereas amplification predominates in embryonal with anaplasia and alveolar subtypes. Genes Chromosomes Cancer. 2002 Mar;33(3):310-21. PMID: #11807989#
![]() Qualman SJ, Coffin CM, Newton WA, Hojo H, Triche TJ, Parham DM, Crist WM. Intergroup Rhabdomyosarcoma Study: update for pathologists. Pediatr Dev Pathol. 1998 Nov-Dec;1(6):550-61. PMID: #9724344#
Qualman SJ, Coffin CM, Newton WA, Hojo H, Triche TJ, Parham DM, Crist WM. Intergroup Rhabdomyosarcoma Study: update for pathologists. Pediatr Dev Pathol. 1998 Nov-Dec;1(6):550-61. PMID: #9724344#