Home > G. Tumoral pathology > paraganglioma
paraganglioma
Thursday 20 November 2003
extra-adrenal pheochromocytomas, phaeochromocytomas; paragangliomas
| PO |
Definition: Paragangliomas arise in specialized neural crest cells associated with segmental or collateral autonomic ganglia throughout the body.
Images
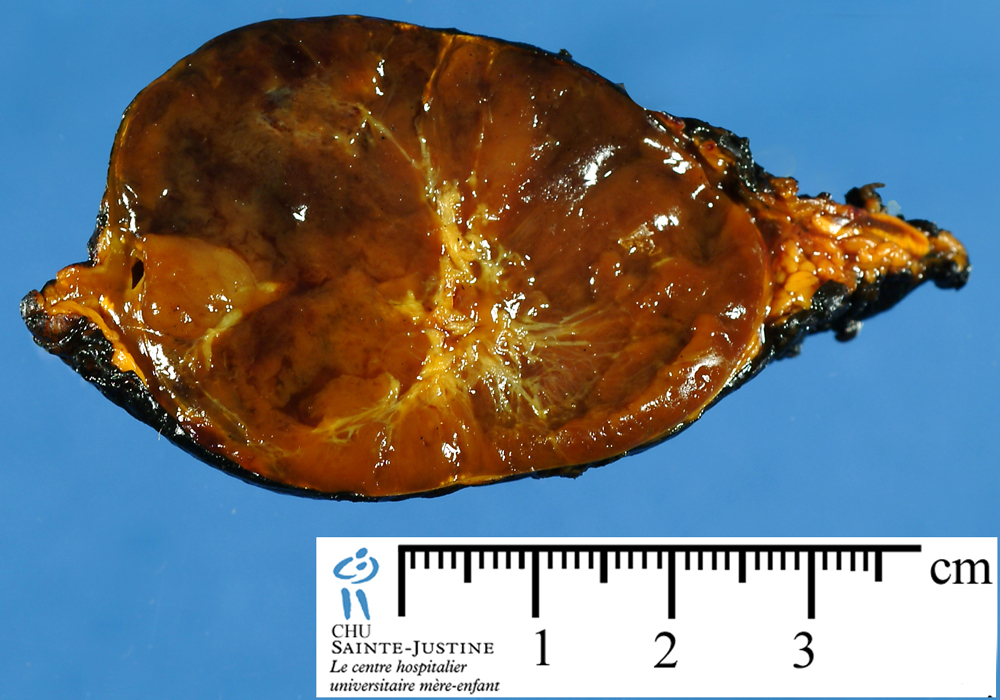
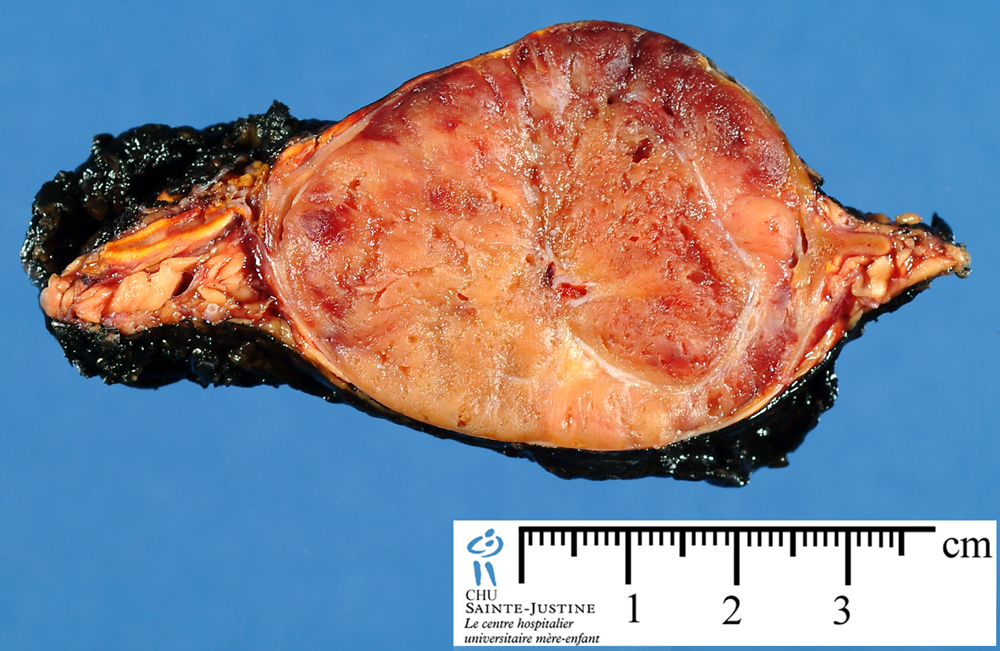
Vagus Paraganglioma
Paraganglioma
- https://twitter.com/raghupillappa/status/816372158066728960
- https://twitter.com/histiocytosisX/status/1147519794046021632
FNA of paragnaglioma
Intranuclear cytoplasmic inclusions in paraganglioma
Digital cases
Case 153 (HPC:153) : Juxtarenal paraganglioma.
Case 331 (HPC:153) : Peritoneum (adrenal) malignant paraganglioma with lymph node metastases.
Extra-adrenal paragangliomas occur most commonly in the head and neck region, usually involving the carotid bodies or glomus jugulare, and are observed less frequently in the mediastinum, retroperitoneum, lungs, duodenum, orbit, larynx, urinary bladder, and spinal cord paragangliomas.
Extra-adrenal phaeochromocytomas occur and may be referred to as paragangliomas, although this term is also used to describe vascular head and neck tumours, which most commonly develop at the carotid bifurcation. Pheochromocytomas are neoplasias of neural crest origin arising from the adrenal medulla.
Paragangliomas (PGLs) are rare neuroendocrine tumors that arise in sympathetic and parasympathetic paraganglia and derive from neural crest cells. Approximately 80–85% of these tumour arise from the adrenal medulla and are named pheochromocytomas (PCCs), whereas 15–20% are located in extra-adrenal chromaffin tissue and are referred to as secreting paragangliomas (sPGLs). The latter term is also used to describe tumors derived from parasympathetic tissue in the head and neck (HNPGLs).
PCCs and abdominal sPGLs are usually catecholamine-producing tumours, whereas most of the HNPGLs are non-functioning.
The majority of PGLs are sporadic, but recent data have demonstrated a high prevalence of hereditary forms (approximately 35%). Sporadic PGLs are usually diagnosed in patients older than 40–50 years, whereas hereditary forms are diagnosed in younger patients.
Malignancy is defined by presence of metastases, tumor spread in sites where chromaffin tissue is normally absent such as lymph nodes, liver, lungs, and bones. Malignant PGLs are extremely rare. An estimated incidence in USA in 2002 was 93 cases per 400 million persons.
Nearly 10% and 20% of PCCs and abdominal sPGLs, respectively, are malignant [4], whereas HNPGLs are usually benign.
Localization
adrenal paraganglioma (pheochromocytoma)
extra-adrenal paraganglioma
- carotid body paraganglioma
- jugulotympanic paraganglioma (glomus jugulare tumor)
- vagal paraganglioma (vagal body tumor)
- mediastinal paraganglioma (aortic body tumor, mediastinal aorticosympathetic paraganglioma)
- retroperitoneal paraganglioma - juxtarenal paraganglioma
- Zuckerkandl organ
- vesical paraganglioma
- cephalic paraganglioma and cervical araganglioma (hean and neck paraganglioma)
- gangliocytic paraganglioma (duodenal, bronchial)
- cauda equina paraganglioma
- nasopharyngeal paraganglioma
- laryngeal paraganglioma
- orbital paraganglioma
- cardiac paraganglioma
- pulmonary paraganglioma (15166677)
- thyroid paraganglioma
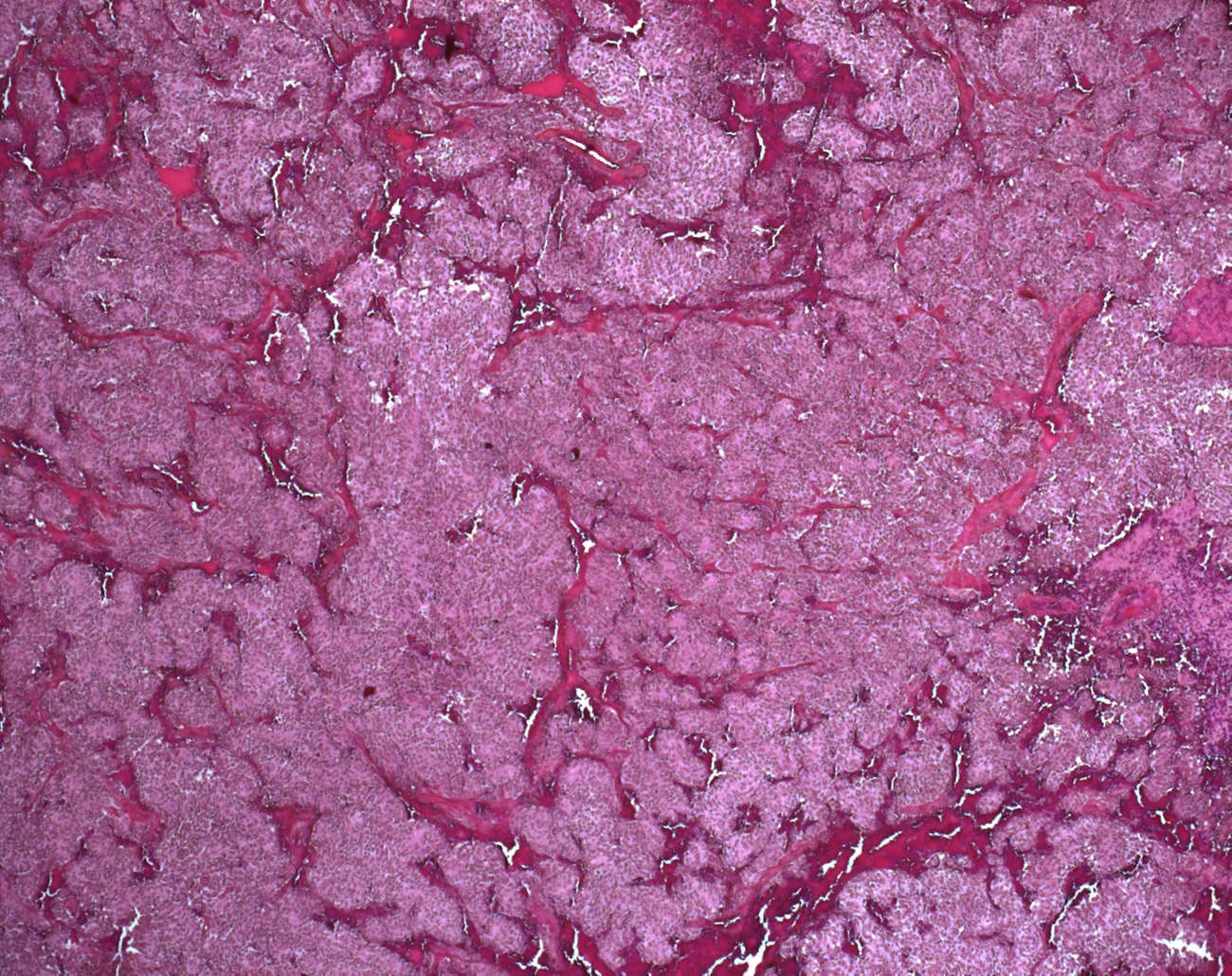
Microscopic synopsis
architectural pattern
- nested architecture
- zellballen architecture/ zellballen pattern
- anastomosing trabecular architecture
- mixed architecture
- compact architecture / diffuse architecture (solid architecture )
- arcuate vascular network
- confluent tumor necrosis
cytological pattern
- argyrophilic cells
- pleomorphic paraganglioma (pleomorphic cells)
- gangliocytic paraganglioma (duodenal)
+/- hyaline globules
mitosis
+/- extensive local invasion
+/- extensive vascular invasion
Variants
composite paraganglioma
pigmented paraganglioma (8491482)
oncocytic pheochromocytoma (11075859)
gangliocytic paraganglioma (15492999)
immature paraganlioma
paraganglioma with extensive gangliocytic differentiation (mixed pheochromocytoma and ganglioneuroma) (3181953, 10839612)
Functional classification
secreting paragngliomas (chromaffin-positive paragangliomas)
non-secreting paragngliomas (chromaffin-negative paragangliomas)
Immunochemistry
NSE+ (100%)
chromogranin-A+
CD56+
synaptophysin+
neurofilament+
Leu-enkephalin+ (76%)
Met-enkephalin+ (75%)
somatostatin+ (67%)
pancreatic polypetide+ (51%)
VIP+ (43%)
substance P+ (31%)
adrenocorticotropic hormon+ (28%)
calcitonin+ (23%)
bombesin+ (15%)
neurotensin+ (12%)
CK- (cytokeratins -)
Differential diagnosis
adrenocortical carcinoma : inhibin+ , MelanA+ and calretinin+
Small blue cell tumors:
- carcinoid tumors / neurendocrine tumors
- Ewing sarcoma / pPNET
- lymphomas
- monomorphic Wilm tumor
- neuroblastoma
- pPNET / medulloblastoma
- rhabdomyosarcomas
- small cell carcinoma - neuroendocrine carcinomas
- small cell osteosarcoma
Ultrastructure
abundant dense core granules with an eccentric halo ("norepinephrine-type" granules)
Susceptibility syndromes
germline mutations may be detected in approximately 25% of unselected cases
von Hippel-Lindau disease (VHL)
multiple endocrine neoplasia type 2 (MEN 2)
phaeochromocytoma-paraganglioma syndrome
neurofibromatosis type 1
familial paraganglioma-GIST syndrome (15383933, 11857563)
Carney triad (paraganglioma, gastrointestinal stromal tumor (GIST), pulmonary chondroma)
SDH mutations / SDH-mutated paraganglioma
- https://twitter.com/CaDxPath/status/1094046992827703296
- Germline mutations in three of the succinate dehydrogenase (SDH, mitochondrial complex II) subunits (SDHD, SDHB and SDHC) cause susceptibility to head and neck paragangliomas, and may be found in approximately 20% of unselected patients.
- In addition, germline SDHD and SDHB mutations may cause phaeochromocytoma susceptibility with or without associated head and neck paragangliomas.
- Recent studies suggest that germline SDHD and SDHB mutations are an important cause of familial and isolated phaeochromocytoma. (dysregulation of hypoxia-responsive genes and impairment of mitochondria-mediated apoptosis)
See "Genetics of paragangliomas"
Prognosis
Despite the increasing availability of molecular diagnostic and prognostic markers, it remains difficult to predict, on the basis of histological findings, whether an apparently benign PGL will develop in a malignant tumor.
From a prognostic point of view, only relative risk factors can be taken into account.
Size
- In general PGLs larger than 5 cm with necrotic areas as well as extra-adrenal tumors carry a higher risk of malignancy than neoplasms that are small or located in the adrenal.
- Several scoring systems considering invasion, histologic growth patterns, cytologic features, or mitotic activity have been proposed to calculate the risk of malignancy.
- One of the most utilized score is “Pheochromocytoma of the Adrenal gland Scales Score (PASS).
- In any event, as none of the available scores predicts malignant development unequivocally, after the removal of an isolated primary PGL, a followup of the patient is recommended in order to reveal early disease recurrence.
- Between histological features, high cellularity and particularly the presence of tumor necrosis are considered potential indicators of malignancy.
Pheochromocytoma of the Adrenal gland Scoring Scale (PASS)
- Nuclear hyperchromasia 1
- Profound nuclear pleomorphism 1
- Capsular invasion 1
- Vascular invasion 1
- Extension into adipose tissue 2
- Atypical mitotic figures 2
- Greater than 3 of 10 mitotic figures high-power field 2
- Tumor cell spindling 2
- Cellular monotony 2
- High cellularity 2
- Central or confluent tumor necrosis 2
- Large nests or diffuse growth (>10% of tumor volume) 2
- Total 20
- A PASS score ≥4 was at first considered suggestive for a biological aggressive behaviour, but a later study revealed that all malignant PCCs had a PASS >6.
- On the basis of these results a PASS score @<@4 or >6 suggest benign and malignant lesions respectively, whereas a value between 4 and 6 suggests an intermediate risk.
Biological prognostic markers
- Several malignancy tissue markers such as cyclooxygenase-2, secretogranin II-derived peptide, N-cadherin, vascular endothelial growth factor (VEGF), endothelin receptor type A (ETA), and type B (ETB) and telomerase have been identified.
- In particular telomerase, which is a ribonucleoprotein complex that includes the telomerase RNA component, the telomerase-associated protein (TP1), the telomerase catalytic subunit (hTERT), and the heat shock protein 90 (HSP90) seem to be closely related to the malignant potential of PGLs.
- In fact an upregulation of hTERT, HSP90, and telomerase activity has been evidenced in malignant cells of PCCs.
Ki67
- The Ki-67 nuclear antigen represents another potential molecular marker which has been associated with more aggressive cancers.
- A Ki-67 index >3% is considered a useful parameter predicting malignant potential.
SNAIL
- Another promising marker predicting metastatic potential seems the transcription factor SNAIL. Positive immunostaining has been found significantly higher in metastatic than benign PGLs.
micro-RNAs (miRNAs)
- Micro-RNA is small single-strand ( 22 bp), nonprotein coding RNA fragments, which are able to negatively regulate protein expression by either cleavage or translational repression of mRNA.
- miR-483-5p is overexpressed, while miR15a and miR-16, which are involved in proliferation and apoptosis, are downregulated in malignant compared to benign tumors.
- MicroRNA expression is tissue specific, and it has been demonstrated to be altered in several other human tumors.
Links
http://www.hindawi.com/journals/jo/2012/872713/
References
Prognosis
V. E. Strong, T. Kennedy, H. Al-Ahmadie et al., “Prognostic indicators of malignancy in adrenal pheochromocytomas: clinical, histopathologic, and cell cycle/apoptosis gene expression analysis,” Surgery, vol. 143, no. 6, pp. 759–768, 2008.
N. Kimura, T. Watanabe, T. Noshiro, S. Shizawa, and Y. Miura, “Histological grading of adrenal and extra-adrenal pheochromocytomas and relationship to prognosis: a clinicopathological analysis of 116 adrenal pheochromocytomas and 30 extra-adrenal sympathetic paragangliomas including 38 malignant tumors,” Endocrine Pathology, vol. 16, no. 1, pp. 23–32, 2005.
T. H. Liu, Y. J. Chen, S. F. Wu et al., “Distinction between benign and malignant pheochromocytomas,” Zhonghua Bing Li Xue Za Zhi, vol. 33, no. 3, pp. 198–202, 2004.
C. Boltze, J. Mundschenk, N. Unger et al., “Expression profile of the telomeric complex discriminates between benign and malignant pheochromocytoma,” Journal of Clinical Endocrinology and Metabolism, vol. 88, no. 9, pp. 4280–4286, 2003.
L. D. R. Thompson, “Pheochromocytoma of the adrenal gland scaled score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases,” American Journal of Surgical Pathology, vol. 26, no. 5, pp. 551–566, 2002.
R. I. Linnoila, H. R. Keiser, S. M. Steinberg, and E. E. Lack, “Histopathology of benign versus malignant sympathoadrenal paragangliomas: clinicopathologic study of 120 cases including unusual histologic features,” Human Pathology, vol. 21, no. 11, pp. 1168–1180, 1990.
Other references
Baysal BE. On the association of succinate dehydrogenase mutations with hereditary paraganglioma. Trends Endocrinol Metab. 2003 Dec;14(10):453-9. PMID: #14643060
Lack EE, Lloyd RV, Carney JA, Woodruff JM; Association of Directors of Anatomic and Surgical Pathology. Recommendations for reporting of extra-adrenal paragangliomas. Mod Pathol. 2003 Aug;16(8):833-5. PMID: 12920230
Lack EE, Lloyd RV, Carney JA, Woodruff JW; Association of Directors of Anatomic and Surgical Pathology. Recommendations for the reporting of extra-adrenal paragangliomas. The Association of Directors of Anatomic and Surgical Pathology. Hum Pathol. 2003 Feb;34(2):112-3. PMID: 12612877
Maher ER, Eng C. The pressure rises: update on the genetics of phaeochromocytoma. Hum Mol Genet. 2002 Oct 1;11(20):2347-54. PMID: 12351569
{kind=link}